The Eukaryotic Translation Initiation Regulator CDC123 Defines A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

BD-CS-057, REV 0 | AUGUST 2017 | Page 1
EXPLIFY RESPIRATORY PATHOGENS BY NEXT GENERATION SEQUENCING Limitations Negative results do not rule out viral, bacterial, or fungal infections. Targeted, PCR-based tests are generally more sensitive and are preferred when specific pathogens are suspected, especially for DNA viruses (Adenovirus, CMV, HHV6, HSV, and VZV), mycobacteria, and fungi. The analytical sensitivity of this test depends on the cellularity of the sample and the concentration of all microbes present. Analytical sensitivity is assessed using Internal Controls that are added to each sample. Sequencing data for Internal Controls is quantified. Samples with Internal Control values below the validated minimum may have reduced analytical sensitivity or contain inhibitors and are reported as ‘Reduced Analytical Sensitivity’. Additional respiratory pathogens to those reported cannot be excluded in samples with ‘Reduced Analytical Sensitivity’. Due to the complexity of next generation sequencing methodologies, there may be a risk of false-positive results. Contamination with organisms from the upper respiratory tract during specimen collection can also occur. The detection of viral, bacterial, and fungal nucleic acid does not imply organisms causing invasive infection. Results from this test need to be interpreted in conjunction with the clinical history, results of other laboratory tests, epidemiologic information, and other available data. Confirmation of positive results by an alternate method may be indicated in select cases. Validated Organisms BACTERIA Achromobacter -

Legionella Shows a Diverse Secondary Metabolism Dependent on a Broad Spectrum Sfp-Type Phosphopantetheinyl Transferase
Legionella shows a diverse secondary metabolism dependent on a broad spectrum Sfp-type phosphopantetheinyl transferase Nicholas J. Tobias1, Tilman Ahrendt1, Ursula Schell2, Melissa Miltenberger1, Hubert Hilbi2,3 and Helge B. Bode1,4 1 Fachbereich Biowissenschaften, Merck Stiftungsprofessur fu¨r Molekulare Biotechnologie, Goethe Universita¨t, Frankfurt am Main, Germany 2 Max von Pettenkofer Institute, Ludwig-Maximilians-Universita¨tMu¨nchen, Munich, Germany 3 Institute of Medical Microbiology, University of Zu¨rich, Zu¨rich, Switzerland 4 Buchmann Institute for Molecular Life Sciences, Goethe Universita¨t, Frankfurt am Main, Germany ABSTRACT Several members of the genus Legionella cause Legionnaires’ disease, a potentially debilitating form of pneumonia. Studies frequently focus on the abundant number of virulence factors present in this genus. However, what is often overlooked is the role of secondary metabolites from Legionella. Following whole genome sequencing, we assembled and annotated the Legionella parisiensis DSM 19216 genome. Together with 14 other members of the Legionella, we performed comparative genomics and analysed the secondary metabolite potential of each strain. We found that Legionella contains a huge variety of biosynthetic gene clusters (BGCs) that are potentially making a significant number of novel natural products with undefined function. Surprisingly, only a single Sfp-like phosphopantetheinyl transferase is found in all Legionella strains analyzed that might be responsible for the activation of all carrier proteins in primary (fatty acid biosynthesis) and secondary metabolism (polyketide and non-ribosomal peptide synthesis). Using conserved active site motifs, we predict Submitted 29 June 2016 some novel compounds that are probably involved in cell-cell communication, Accepted 25 October 2016 Published 24 November 2016 differing to known communication systems. -

The Value of UV for Legionella Control in Cooling Towers
The Value of UV for Legionella Control in Cooling Towers By Ytzhak Rozenberg, Assaf Lowentahl, Ph.D. Atlantium Technologies, [email protected], www.atlantium.com. ABSTRACT: Excessive growth of Legionella in cooling towers and water systems can cause significant negative health effects. Legionella and free-living amoebae may be present together and amoebae can act as a shield for Legionella, protecting it from traditional chemical disinfectants. Ultraviolet (UV) disinfection offers a non-chemical treatment approach that is effective for reducing biofilm potential in cooling tower water and provides reliable protection against the spread of Legionella and inac- tivation of amoebae. However, all UV technologies are not created equal; low-pressure (LP) and medium-pressure (MP) UV lamp technology have different effects on a microorganism’s mortality rate at different UV dose rates. In this comparative study, a proprietary MP lamp technology (Hydro-Optic™ [HOD]) manufactured by Atlantium Technologies was evaluated and determined to achieve 100% mortality of Entamoeba Histolytica (E. histolytica) at a dose of 8.8 mJ/cm2, while LP UV systems, even at a dose of 90 mJ/cm2, did not achieve 100% mortality. E. histolytica served as a Legionella host model in the study. INTRODUCTION Cooling towers operate at temperatures that can provide optimal conditions for the growth of microorganisms in wa- Various studies have shown that 40-60% of all cooling tow- ter (20-45°C, 68-113°F), including Legionella. ers harbor Legionella bacteria. Cooling towers are the larg- est and most common source of Legionnaire’s disease out- Other operating conditions contributing to the growth of Le- breaks because of their risk for widespread circulation. -

Genetic and Functional Studies of the Mip Protein of Legionella
t1.ì. Genetic and Functional Studies of the Mip Protein of Legionella Rodney Mark Ratcliff, BSc (Hons)' MASM Infectious Diseases Laboratories Institute of Medical and Veterinary Science and Department of Microbiology and Immunology UniversitY of Adelaide. Adelaide, South Australia A thesis submitted to the University of Adelaide for the degree of I)octor of Philosophy 15'h March 2000 amended 14th June 2000 Colonies of several Legionella strains on charcoal yeast extract agar (CYE) after 4 days incubation at 37"C in air. Various magnifications show typical ground-glass opalescent appearance. Some pure strains exhibit pleomorphic growth or colour. The top two photographs demonstrate typical red (LH) and blue-white (RH) fluorescence exhibited by some species when illuminated by a Woods (IJV) Lamp. * t Table of Contents .1 Chapter One: Introduction .1 Background .............'. .2 Morphology and TaxonomY J Legionellosis ............. 5 Mode of transmission "..'....'. 7 Environmental habitat 8 Interactions between Legionella and phagocytic hosts 9 Attachment 11 Engulfment and internalisation.'.. 13 Intracellular processing 13 Intracellular replication of legionellae .. " "' " "' 15 Host cell death and bacterial release 18 Virulence (the Genetic factors involved with intracellular multiplication and host cell killing .20 icm/dot system) Legiolysin .25 Msp (Znn* metaloprotease) ...'..... .25 .28 Lipopolysaccharide .29 The association of flagella with disease.. .30 Type IV fimbriae.... .31 Major outer membrane proteins....'.......'. JJ Heat shock proteins'.'. .34 Macrophage infectivity potentiator (Mip) protein Virulenceiraits of Legionella species other than L. pneumophila..........' .39 phylogeny .41 Chapter One (continued): Introduction to bacterial classification and .41 Identificati on of Legionella...'.,..'.. .46 Phylogeny .52 Methods of phylogenetic analysis' .53 Parsimony methods.'.. .55 Distance methods UPGMA cluster analYsis.'.'... -

EVALUATION of the P45 MOBILE INTEGRATIVE ELEMENT and ITS ROLE IN
EVALUATION OF THE p45 MOBILE INTEGRATIVE ELEMENT AND ITS ROLE IN Legionella pneumophila VIRULENCE A Dissertation by LANETTE M. CHRISTENSEN Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Chair of Committee, Jeffrey D. Cirillo Committee Members, James Samuel Jon Skare Farida Sohrabji Head of Program, Warren Zimmer May 2018 Major Subject: Medical Sciences Copyright 2018 Lanette Christensen ABSTRACT Legionella pneumophila are aqueous environmental bacilli that live within protozoal species and cause a potentially fatal form of pneumonia called Legionnaires’ disease. Not all L. pneumophila strains have the same capacity to cause disease in humans. The majority of strains that cause clinically relevant Legionnaires’ disease harbor the p45 mobile integrative genomic element. Contribution of the p45 element to L. pneumophila virulence and ability to withstand environmental stress were addressed in this study. The L. pneumophila Philadelphia-1 (Phil-1) mobile integrative element, p45, was transferred into the attenuated strain Lp01 via conjugation, designating p45 an integrative conjugative element (ICE). The resulting trans-conjugate, Lp01+p45, was compared with strains Phil-1 and Lp01 to assess p45 in virulence using a guinea pig model infected via aerosol. The p45 element partially recovered the loss of virulence in Lp01 compared to that of Phil-1 evident in morbidity, mortality, and bacterial burden in the lungs at the time of death. This phenotype was accompanied by enhanced expression of type II interferon in the lungs and spleens 48 hours after infection, independent of bacterial burden. -
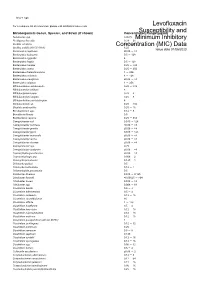
Susceptibility and Resistance Data
toku-e logo For a complete list of references, please visit antibiotics.toku-e.com Levofloxacin Microorganism Genus, Species, and Strain (if shown) Concentration Range (μg/ml)Susceptibility and Aeromonas spp. 0.0625 Minimum Inhibitory Alcaligenes faecalis 0.39 - 25 Bacillus circulans Concentration0.25 - 8 (MIC) Data Bacillus subtilis (ATCC 6051) 6.25 Issue date 01/06/2020 Bacteroides capillosus ≤0.06 - >8 Bacteroides distasonis 0.5 - 128 Bacteroides eggerthii 4 Bacteroides fragilis 0.5 - 128 Bacteroides merdae 0.25 - >32 Bacteroides ovatus 0.25 - 256 Bacteroides thetaiotaomicron 1 - 256 Bacteroides uniformis 4 - 128 Bacteroides ureolyticus ≤0.06 - >8 Bacteroides vulgatus 1 - 256 Bifidobacterium adolescentis 0.25 - >32 Bifidobacterium bifidum 8 Bifidobacterium breve 0.25 - 8 Bifidobacterium longum 0.25 - 8 Bifidobacterium pseudolongum 8 Bifidobacterium sp. 0.25 - >32 Bilophila wadsworthia 0.25 - 16 Brevibacterium spp. 0.12 - 8 Brucella melitensis 0.5 Burkholderia cepacia 0.25 - 512 Campylobacter coli 0.015 - 128 Campylobacter concisus ≤0.06 - >8 Campylobacter gracilis ≤0.06 - >8 Campylobacter jejuni 0.015 - 128 Campylobacter mucosalis ≤0.06 - >8 Campylobacter rectus ≤0.06 - >8 Campylobacter showae ≤0.06 - >8 Campylobacter spp. 0.25 Campylobacter sputorum ≤0.06 - >8 Capnocytophaga ochracea ≤0.06 - >8 Capnocytophaga spp. 0.006 - 2 Chlamydia pneumonia 0.125 - 1 Chlamydia psittaci 0.5 Chlamydia trachomatis 0.12 - 1 Chlamydophila pneumonia 0.5 Citrobacter diversus 0.015 - 0.125 Citrobacter freundii ≤0.00625 - >64 Citrobacter koseri 0.015 - -

Aquascreen® Legionella Species Qpcr Detection Kit
AquaScreen® Legionella species qPCR Detection Kit INSTRUCTIONS FOR USE FOR USE IN RESEARCH AND QUALITY CONTROL Symbols Lot No. Cat. No. Expiry date Storage temperature Number of reactions Manufacturer INDICATION The AquaScreen® Legionella species qPCR Detection kit is specifically designed for the quantitative detection of several Legionella species in water samples prepared with the AquaScreen® FastExt- ract kit. Its design complies with the requirements of AFNOR T90-471 and ISO/TS 12869:2012. Legionella are ubiquitous bacteria in surface water and moist soil, where they parasitize protozoa. The optimal growth temperature lies between +15 and +45 °C, whereas these gram-negative bacteria are dormant below 20 °C and do not survive above 60 °C. Importantly, Legionella are well-known as opportunistic intracellular human pathogens causing Legionnaires’ disease and Pontiac fever. The transmission occurs through inhalation of contami- nated aerosols generated by an infected source (e.g. human-made water systems like shower- heads, sink faucets, heaters, cooling towers, and many more). In order to efficiently prevent Legionella outbreaks, water safety control measures need syste- matic application but also reliable validation by fast Legionella testing. TEST PRINCIPLE The AquaScreen® Legionella species Kit uses qPCR for quantitative detection of legionella in wa- ter samples. In contrast to more time-consuming culture-based methods, AquaScreen® assays need less than six hours including sample preparation and qPCR to reliably detect Legionella. Moreover, the AquaScreen® qPCR assay has proven excellent performance in terms of specificity and sensitivity: other bacterial genera remain undetected whereas linear quantification is obtai- ned up to 1 x 106 particles per sample, therefore requiring no material dilution. -

Neocallimastix Californiae G1 36,250,970 NA 29,649 95.52 85.2 SRX2598479 (3)
Supplementary material for: Horizontal gene transfer as an indispensable driver for Neocallimastigomycota evolution into a distinct gut-dwelling fungal lineage 1 1 1 2 Chelsea L. Murphy ¶, Noha H. Youssef ¶, Radwa A. Hanafy , MB Couger , Jason E. Stajich3, Y. Wang3, Kristina Baker1, Sumit S. Dagar4, Gareth W. Griffith5, Ibrahim F. Farag1, TM Callaghan6, and Mostafa S. Elshahed1* Table S1. Validation of HGT-identification pipeline using previously published datasets. The frequency of HGT occurrence in the genomes of a filamentous ascomycete and a microsporidian were determined using our pipeline. The results were compared to previously published results. Organism NCBI Assembly Reference Method used Value Value accession number to original in the original reported obtained study study in this study Colletotrichum GCA_000149035.1 (1) Blast and tree 11 11 graminicola building approaches Encephalitozoon GCA_000277815.3 (2) Blast against 12-22 4 hellem custom database, AI score calculation, and tree building Table S2. Results of transcriptomic sequencing. Accession number Genus Species Strain Number of Assembled Predicted peptides % genome Ref. reads transcriptsa (Longest Orfs)b completenessc coveraged (%) Anaeromyces contortus C3G 33,374,692 50,577 22,187 96.55 GGWR00000000 This study Anaeromyces contortus C3J 54,320,879 57,658 26,052 97.24 GGWO00000000 This study Anaeromyces contortus G3G 43,154,980 52,929 21,681 91.38 GGWP00000000 This study Anaeromyces contortus Na 42,857,287 47,378 19,386 93.45 GGWN00000000 This study Anaeromyces contortus O2 60,442,723 62,300 27,322 96.9 GGWQ00000000 This study Anaeromyces robustus S4 21,955,935 NA 17,127 92.41 88.7 SRX3329608 (3) Caecomyces sp. -

Comparative Analyses of Legionella Species Identifies Genetic Features
Gomez-Valero et al. Genome Biology 2014, 15:505 http://genomebiology.com/2014/15/11/505 RESEARCH Open Access Comparative analyses of Legionella species identifies genetic features of strains causing Legionnaires’ disease Laura Gomez-Valero1,2*, Christophe Rusniok1,2, Monica Rolando1,2, Mario Neou1,2, Delphine Dervins-Ravault1,2, Jasmin Demirtas1,2, Zoe Rouy9, Robert J Moore10, Honglei Chen10, Nicola K Petty3, Sophie Jarraud4,5, Jerome Etienne4,5, Michael Steinert6, Klaus Heuner7, Simonetta Gribaldo8, Claudine Médigue9, Gernot Glöckner11, Elizabeth L Hartland12 and Carmen Buchrieser1* Abstract Background: The genus Legionella comprises over 60 species. However, L. pneumophila and L. longbeachae alone cause over 95% of Legionnaires’ disease. To identify the genetic bases underlying the different capacities to cause disease we sequenced and compared the genomes of L. micdadei, L. hackeliae and L. fallonii (LLAP10), which are all rarely isolated from humans. Results: We show that these Legionella species possess different virulence capacities in amoeba and macrophages, correlating with their occurrence in humans. Our comparative analysis of 11 Legionella genomes belonging to five species reveals highly heterogeneous genome content with over 60% representing species-specific genes; these comprise a complete prophage in L. micdadei, the first ever identified in a Legionella genome. Mobile elements are abundant in Legionella genomes; many encode type IV secretion systems for conjugative transfer, pointing to their importance for adaptation of the genus. The Dot/Icm secretion system is conserved, although the core set of substrates is small, as only 24 out of over 300 described Dot/Icm effector genes are present in all Legionella species. We also identified new eukaryotic motifs including thaumatin, synaptobrevin or clathrin/coatomer adaptine like domains. -

HL7 Implementation Guide for CDA Release 2: NHSN Healthcare Associated Infection (HAI) Reports, Release 2 (U.S
CDAR2L3_IG_HAIRPT_R2_D2_2009FEB HL7 Implementation Guide for CDA Release 2: NHSN Healthcare Associated Infection (HAI) Reports, Release 2 (U.S. Realm) Draft Standard for Trial Use February 2009 © 2009 Health Level Seven, Inc. Ann Arbor, MI All rights reserved HL7 Implementation Guide for CDA R2 NHSN HAI Reports Page 1 Draft Standard for Trial Use Release 2 February 2009 © 2009 Health Level Seven, Inc. All rights reserved. Co-Editor: Marla Albitz Contractor to CDC, Lockheed Martin [email protected] Co-Editor/Co-Chair: Liora Alschuler Alschuler Associates, LLC [email protected] Co-Chair: Calvin Beebe Mayo Clinic [email protected] Co-Chair: Keith W. Boone GE Healthcare [email protected] Co-Editor/Co-Chair: Robert H. Dolin, M.D. Semantically Yours, LLC [email protected] Co-Editor Jonathan Edwards CDC [email protected] Co-Editor Pavla Frazier RN, MSN, MBA Contractor to CDC, Frazier Consulting [email protected] Co-Editor: Sundak Ganesan, M.D. SAIC Consultant to CDC/NCPHI [email protected] Co-Editor: Rick Geimer Alschuler Associates, LLC [email protected] Co-Editor: Gay Giannone M.S.N., R.N. Alschuler Associates LLC [email protected] Co-Editor Austin Kreisler Consultant to CDC/NCPHI [email protected] Primary Editor: Kate Hamilton Alschuler Associates, LLC [email protected] Primary Editor: Brett Marquard Alschuler Associates, LLC [email protected] Co-Editor: Daniel Pollock, M.D. CDC [email protected] HL7 Implementation Guide for CDA R2 NHSN HAI Reports Page 2 Draft Standard for Trial Use Release 2 February 2009 © 2009 Health Level Seven, Inc. All rights reserved. Acknowledgments This DSTU was produced and developed in conjunction with the Division of Healthcare Quality Promotion, National Center for Preparedness, Detection, and Control of Infectious Diseases (NCPDCID), and Centers for Disease Control and Prevention (CDC). -

PROTEINS of Legionella Pneumophila
IMMUNE RESPONSE OF THE RAT TO OUTER MEMBRANE PROTEINS OF Legionella pneumophila APPROVED: 1%.iGraduate " %*% %-% %.oN.0 Committee: Maj r Prr essqx Minor Professor/ / Committee Member Committ.96 Member Comnnrttde M6mber Chairperson of the Department of Biological Sciences Dean of the G'adhlte Scho I WON 37? /d8 /ci IMMUNE RESPONSE OF THE RAT TO OUTER MEMBRANE PROTEINS OF Legionella pneumophila DISSERTATION Presented to the Graduate Council of the North Texas State University in Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY By Ejemihu Ndu Ahanotu, M. A. Denton, Texas August, 1985 Ahanotu, Ndu Ejemihu, Immune Response of the Rat to Outer Membrane Proteins of Legionella pneumophila. Doctor of Philosophy (Microbiology), August, 1985, 163 pp., 14 tables, bibliography, 225 titles. Outer membrane proteins (OMPs) were recovered from eleven strains (eight serogroups) of Legionella pneumophila by sequential treatment with Tris buffer (pH 8), citrate buffer (pH 2.75) and Tris buffer (pH 8). Transmission electron microscopy revealed clearly the separation of the outer membrane from the bacteria. The solubilized OMPs, separated and visualized by discontinuous polyacrylamide gel electrophoresis, contained a large variety of proteins ranging from 10 to 79 Kd. A 24 Kd band was common to all eight serogroups. A male New Zealand white rabbit infected with 1.5 x 106 L. pneumophila (Chicago 8, serogroup 7) died in nine days despite the administration of erythromycin. Histopathology was particularly evident in the heart and lungs. Both myocardial lesions and pneumonitis were evident. Active immunity was produced in rats injected in the footpads with the OMPs from Chicago 8 strain employing Freud's com- plete adjuvant, a double emulsion adjuvant or saline. -

Legionella Feeleii: Pneumonia Or Pontiac Fever? Bacterial Virulence Traits and Host Immune Response
Medical Microbiology and Immunology (2019) 208:25–32 https://doi.org/10.1007/s00430-018-0571-0 REVIEW Legionella feeleii: pneumonia or Pontiac fever? Bacterial virulence traits and host immune response Changle Wang1 · Xia Chuai1 · Mei Liang2 Received: 17 July 2018 / Accepted: 27 October 2018 / Published online: 1 November 2018 © Springer-Verlag GmbH Germany, part of Springer Nature 2018 Abstract Gram-negative bacterium Legionella is able to proliferate intracellularly in mammalian host cells and amoeba, which became known in 1976 since they caused a large outbreak of pneumonia. It had been reported that different strains of Legionella pneumophila, Legionella micdadei, Legionella longbeachae, and Legionella feeleii caused human respiratory diseases, which were known as Pontiac fever or Legionnaires’ disease. However, the differences of the virulence traits among the strains of the single species and the pathogenesis of the two diseases that were due to the bacterial virulence factors had not been well elucidated. L. feeleii is an important pathogenic organism in Legionellae, which attracted attention due to cause an outbreak of Pontiac fever in 1981 in Canada. In published researches, it has been found that L. feeleii serogroup 2 (ATCC 35849, LfLD) possess mono-polar flagellum, and L. feeleii serogroup 1 (ATCC 35072, WRLf) could secrete some exopolysaccharide (EPS) materials to the surrounding. Although the virulence of the L. feeleii strain was evidenced that could be promoted, the EPS might be dispensable for the bacteria that caused Pontiac fever. Based on the current knowledge, we focused on bacterial infection in human and murine host cells, intracellular growth, cytopathogenicity, stimulatory capacity of cytokines secre- tion, and pathogenic effects of the EPS ofL.