Replication Stress Leading to Apoptosis Within the S-Phase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

By Submitted in Partial Satisfaction of the Requirements for Degree of in In
Developments of Two Imaging based Technologies for Cell Biology Researches by Xiaowei Yan DISSERTATION Submitted in partial satisfaction of the requirements for degree of DOCTOR OF PHILOSOPHY in Biochemistry and Molecular Biology in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO Approved: ______________________________________________________________________________Ronald Vale Chair ______________________________________________________________________________Jonathan Weissman ______________________________________________________________________________Orion Weiner ______________________________________________________________________________ ______________________________________________________________________________ Committee Members Copyright 2021 By Xiaowei Yan ii DEDICATION Everything happens for the best. To my family, who supported me with all their love. iii ACKNOWLEDGEMENTS The greatest joy of my PhD has been joining UCSF, working and learning with such a fantastic group of scientists. I am extremely grateful for all the support and mentorship I received and would like to thank: My mentor, Ron Vale, who is such a great and generous person. Thank you for showing me that science is so much fun and thank you for always giving me the freedom in pursuing my interest. I am grateful for all the guidance from you and thank you for always supporting me whenever I needed. You are a person full of wisdom, and I have been learning so much from you and your attitude to science, science community and even life will continue inspire me. Thank you for being my mentor and thank you for being such a great mentor. Everyone else in Vale lab, past and present, for making our lab a sweet home. I would like to give my special thank to Marvin (Marvin Tanenbaum) and Nico (Nico Stuurman), two other mentors for me in the lab. I would like to thank them for helping me adapt to our lab, for all the valuable advice and for all the happiness during the time that we work together. -

Nuclear Organization and the Epigenetic Landscape of the Mus Musculus X-Chromosome Alicia Liu University of Connecticut - Storrs, [email protected]
University of Connecticut OpenCommons@UConn Doctoral Dissertations University of Connecticut Graduate School 8-9-2019 Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome Alicia Liu University of Connecticut - Storrs, [email protected] Follow this and additional works at: https://opencommons.uconn.edu/dissertations Recommended Citation Liu, Alicia, "Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome" (2019). Doctoral Dissertations. 2273. https://opencommons.uconn.edu/dissertations/2273 Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome Alicia J. Liu, Ph.D. University of Connecticut, 2019 ABSTRACT X-linked imprinted genes have been hypothesized to contribute parent-of-origin influences on social cognition. A cluster of imprinted genes Xlr3b, Xlr4b, and Xlr4c, implicated in cognitive defects, are maternally expressed and paternally silent in the murine brain. These genes defy classic mechanisms of autosomal imprinting, suggesting a novel method of imprinted gene regulation. Using Xlr3b and Xlr4c as bait, this study uses 4C-Seq on neonatal whole brain of a 39,XO mouse model, to provide the first in-depth analysis of chromatin dynamics surrounding an imprinted locus on the X-chromosome. Significant differences in long-range contacts exist be- tween XM and XP monosomic samples. In addition, XM interaction profiles contact a greater number of genes linked to cognitive impairment, abnormality of the nervous system, and abnormality of higher mental function. This is not a pattern that is unique to the imprinted Xlr3/4 locus. Additional Alicia J. Liu - University of Connecticut - 2019 4C-Seq experiments show that other genes on the X-chromosome, implicated in intellectual disability and/or ASD, also produce more maternal contacts to other X-linked genes linked to cognitive impairment. -

Gene Knockdown of CENPA Reduces Sphere Forming Ability and Stemness of Glioblastoma Initiating Cells
Neuroepigenetics 7 (2016) 6–18 Contents lists available at ScienceDirect Neuroepigenetics journal homepage: www.elsevier.com/locate/nepig Gene knockdown of CENPA reduces sphere forming ability and stemness of glioblastoma initiating cells Jinan Behnan a,1, Zanina Grieg b,c,1, Mrinal Joel b,c, Ingunn Ramsness c, Biljana Stangeland a,b,⁎ a Department of Molecular Medicine, Institute of Basic Medical Sciences, The Medical Faculty, University of Oslo, Oslo, Norway b Norwegian Center for Stem Cell Research, Department of Immunology and Transfusion Medicine, Oslo University Hospital, Oslo, Norway c Vilhelm Magnus Laboratory for Neurosurgical Research, Institute for Surgical Research and Department of Neurosurgery, Oslo University Hospital, Oslo, Norway article info abstract Article history: CENPA is a centromere-associated variant of histone H3 implicated in numerous malignancies. However, the Received 20 May 2016 role of this protein in glioblastoma (GBM) has not been demonstrated. GBM is one of the most aggressive Received in revised form 23 July 2016 human cancers. GBM initiating cells (GICs), contained within these tumors are deemed to convey Accepted 2 August 2016 characteristics such as invasiveness and resistance to therapy. Therefore, there is a strong rationale for targeting these cells. We investigated the expression of CENPA and other centromeric proteins (CENPs) in Keywords: fi CENPA GICs, GBM and variety of other cell types and tissues. Bioinformatics analysis identi ed the gene signature: fi Centromeric proteins high_CENP(AEFNM)/low_CENP(BCTQ) whose expression correlated with signi cantly worse GBM patient Glioblastoma survival. GBM Knockdown of CENPA reduced sphere forming ability, proliferation and cell viability of GICs. We also Brain tumor detected significant reduction in the expression of stemness marker SOX2 and the proliferation marker Glioblastoma initiating cells and therapeutic Ki67. -
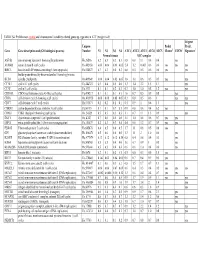
TABLE S4: Proliferation-Related and Chromosomal Instability-Related
TABLE S4: Proliferation-related and chromosomal instability-related genes up-regulated in ATC (weight <4.0) 44-gene Unigene Prolif. Prolif. Gene Gene description and (GO biological process) Number N1 N2 N3 N4 ATC1 ATC2 ATC3 ATC4 ATC5 Cluster1 CIN702 Signature3 Normal tissues ATC samples ASF1B anti-silencing function 1 homolog B (unknown) Hs.26516 0.2 0.3 0.3 0.3 0.8 0.8 1.1 0.8 0.4 yes AURKB aurora kinase B (cell cycle) Hs.442658 0.06 0.06 0.08 0.05 2.4 1.2 0.003 0.6 0.6 yes yes yes BIRC5 baculoviral IAP repeat-containing 5 (anti apoptosis) Hs.514527 0.7 0.3 0.4 0.3 0.8 0.5 0.5 0.6 0.6 yes yes budding uninhibited by benzimidazoles 1 homolog (mitotic BUB1 spindle checkpoint) Hs.469649 0.06 0.04 0.02 0.05 1.8 1.0 0.6 0.5 0.5 yes yes CCNE1 cyclin E1 (cell cycle) Hs.244723 0.5 0.4 0.5 0.6 1.3 1.4 2.2 1.5 1.1 yes CCNF cyclin F (cell cycle) Hs.1973 0.1 0.1 0.2 0.3 0.7 1.0 1.0 0.8 1.2 yes yes CDC45L CDC45 cell division cycle 45-like (cell cycle) Hs.474217 0.1 0.1 0.1 0.3 1.6 0.7 0.5 0.9 0.8 yes CDC6 cell division cycle 6 homolog (cell cycle) Hs. 405958 0.08 0.08 0.08 0.07 0.3 0.8 0.5 0.6 1 yes yes CDC7 cell division cycle 7 (cell cycle) Hs.533573 0.2 0.2 0.2 0.1 0.5 0.9 1 0.6 1.3 yes CDKN3 cyclin-dependent kinase inhibitor 3 (cell cycle) Hs.84113 0.1 0.1 0.7 0.1 0.9 0.8 0.6 0.4 6.2 yes CHEK1 CHK1 checkpoint homolog (cell cycle) Hs. -

Identification of Conserved Genes Triggering Puberty in European Sea
Blázquez et al. BMC Genomics (2017) 18:441 DOI 10.1186/s12864-017-3823-2 RESEARCHARTICLE Open Access Identification of conserved genes triggering puberty in European sea bass males (Dicentrarchus labrax) by microarray expression profiling Mercedes Blázquez1,2* , Paula Medina1,2,3, Berta Crespo1,4, Ana Gómez1 and Silvia Zanuy1* Abstract Background: Spermatogenesisisacomplexprocesscharacterized by the activation and/or repression of a number of genes in a spatio-temporal manner. Pubertal development in males starts with the onset of the first spermatogenesis and implies the division of primary spermatogonia and their subsequent entry into meiosis. This study is aimed at the characterization of genes involved in the onset of puberty in European sea bass, and constitutes the first transcriptomic approach focused on meiosis in this species. Results: European sea bass testes collected at the onset of puberty (first successful reproduction) were grouped in stage I (resting stage), and stage II (proliferative stage). Transition from stage I to stage II was marked by an increase of 11ketotestosterone (11KT), the main fish androgen, whereas the transcriptomic study resulted in 315 genes differentially expressed between the two stages. The onset of puberty induced 1) an up-regulation of genes involved in cell proliferation, cell cycle and meiosis progression, 2) changes in genes related with reproduction and growth, and 3) a down-regulation of genes included in the retinoic acid (RA) signalling pathway. The analysis of GO-terms and biological pathways showed that cell cycle, cell division, cellular metabolic processes, and reproduction were affected, consistent with the early events that occur during the onset of puberty. -

Supplementary Table S5. Differentially Expressed Gene Lists of PD-1High CD39+ CD8 Tils According to 4-1BB Expression Compared to PD-1+ CD39- CD8 Tils
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Immunother Cancer Supplementary Table S5. Differentially expressed gene lists of PD-1high CD39+ CD8 TILs according to 4-1BB expression compared to PD-1+ CD39- CD8 TILs Up- or down- regulated genes in Up- or down- regulated genes Up- or down- regulated genes only PD-1high CD39+ CD8 TILs only in 4-1BBneg PD-1high CD39+ in 4-1BBpos PD-1high CD39+ CD8 compared to PD-1+ CD39- CD8 CD8 TILs compared to PD-1+ TILs compared to PD-1+ CD39- TILs CD39- CD8 TILs CD8 TILs IL7R KLRG1 TNFSF4 ENTPD1 DHRS3 LEF1 ITGA5 MKI67 PZP KLF3 RYR2 SIK1B ANK3 LYST PPP1R3B ETV1 ADAM28 H2AC13 CCR7 GFOD1 RASGRP2 ITGAX MAST4 RAD51AP1 MYO1E CLCF1 NEBL S1PR5 VCL MPP7 MS4A6A PHLDB1 GFPT2 TNF RPL3 SPRY4 VCAM1 B4GALT5 TIPARP TNS3 PDCD1 POLQ AKAP5 IL6ST LY9 PLXND1 PLEKHA1 NEU1 DGKH SPRY2 PLEKHG3 IKZF4 MTX3 PARK7 ATP8B4 SYT11 PTGER4 SORL1 RAB11FIP5 BRCA1 MAP4K3 NCR1 CCR4 S1PR1 PDE8A IFIT2 EPHA4 ARHGEF12 PAICS PELI2 LAT2 GPRASP1 TTN RPLP0 IL4I1 AUTS2 RPS3 CDCA3 NHS LONRF2 CDC42EP3 SLCO3A1 RRM2 ADAMTSL4 INPP5F ARHGAP31 ESCO2 ADRB2 CSF1 WDHD1 GOLIM4 CDK5RAP1 CD69 GLUL HJURP SHC4 GNLY TTC9 HELLS DPP4 IL23A PITPNC1 TOX ARHGEF9 EXO1 SLC4A4 CKAP4 CARMIL3 NHSL2 DZIP3 GINS1 FUT8 UBASH3B CDCA5 PDE7B SOGA1 CDC45 NR3C2 TRIB1 KIF14 TRAF5 LIMS1 PPP1R2C TNFRSF9 KLRC2 POLA1 CD80 ATP10D CDCA8 SETD7 IER2 PATL2 CCDC141 CD84 HSPA6 CYB561 MPHOSPH9 CLSPN KLRC1 PTMS SCML4 ZBTB10 CCL3 CA5B PIP5K1B WNT9A CCNH GEM IL18RAP GGH SARDH B3GNT7 C13orf46 SBF2 IKZF3 ZMAT1 TCF7 NECTIN1 H3C7 FOS PAG1 HECA SLC4A10 SLC35G2 PER1 P2RY1 NFKBIA WDR76 PLAUR KDM1A H1-5 TSHZ2 FAM102B HMMR GPR132 CCRL2 PARP8 A2M ST8SIA1 NUF2 IL5RA RBPMS UBE2T USP53 EEF1A1 PLAC8 LGR6 TMEM123 NEK2 SNAP47 PTGIS SH2B3 P2RY8 S100PBP PLEKHA7 CLNK CRIM1 MGAT5 YBX3 TP53INP1 DTL CFH FEZ1 MYB FRMD4B TSPAN5 STIL ITGA2 GOLGA6L10 MYBL2 AHI1 CAND2 GZMB RBPJ PELI1 HSPA1B KCNK5 GOLGA6L9 TICRR TPRG1 UBE2C AURKA Leem G, et al. -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -

Supplementary Table S1. Correlation Between the Mutant P53-Interacting Partners and PTTG3P, PTTG1 and PTTG2, Based on Data from Starbase V3.0 Database
Supplementary Table S1. Correlation between the mutant p53-interacting partners and PTTG3P, PTTG1 and PTTG2, based on data from StarBase v3.0 database. PTTG3P PTTG1 PTTG2 Gene ID Coefficient-R p-value Coefficient-R p-value Coefficient-R p-value NF-YA ENSG00000001167 −0.077 8.59e-2 −0.210 2.09e-6 −0.122 6.23e-3 NF-YB ENSG00000120837 0.176 7.12e-5 0.227 2.82e-7 0.094 3.59e-2 NF-YC ENSG00000066136 0.124 5.45e-3 0.124 5.40e-3 0.051 2.51e-1 Sp1 ENSG00000185591 −0.014 7.50e-1 −0.201 5.82e-6 −0.072 1.07e-1 Ets-1 ENSG00000134954 −0.096 3.14e-2 −0.257 4.83e-9 0.034 4.46e-1 VDR ENSG00000111424 −0.091 4.10e-2 −0.216 1.03e-6 0.014 7.48e-1 SREBP-2 ENSG00000198911 −0.064 1.53e-1 −0.147 9.27e-4 −0.073 1.01e-1 TopBP1 ENSG00000163781 0.067 1.36e-1 0.051 2.57e-1 −0.020 6.57e-1 Pin1 ENSG00000127445 0.250 1.40e-8 0.571 9.56e-45 0.187 2.52e-5 MRE11 ENSG00000020922 0.063 1.56e-1 −0.007 8.81e-1 −0.024 5.93e-1 PML ENSG00000140464 0.072 1.05e-1 0.217 9.36e-7 0.166 1.85e-4 p63 ENSG00000073282 −0.120 7.04e-3 −0.283 1.08e-10 −0.198 7.71e-6 p73 ENSG00000078900 0.104 2.03e-2 0.258 4.67e-9 0.097 3.02e-2 Supplementary Table S2. -

Non-Synonymous Mutations Mapped to Chromosome X Associated With
de Camargo et al. BMC Genomics (2015) 16:384 DOI 10.1186/s12864-015-1595-0 RESEARCH ARTICLE Open Access Non-synonymous mutations mapped to chromosome X associated with andrological and growth traits in beef cattle Gregório Miguel Ferreira de Camargo1,2,3, Laercio R Porto-Neto2, Matthew J Kelly3, Rowan J Bunch2, Sean M McWilliam2, Humberto Tonhati1, Sigrid A Lehnert2, Marina R S Fortes3* and Stephen S Moore4 Abstract Background: Previous genome-wide association analyses identified QTL regions in the X chromosome for percentage of normal sperm and scrotal circumference in Brahman and Tropical Composite cattle. These traits are important to be studied because they are indicators of male fertility and are correlated with female sexual precocity and reproductive longevity. The aim was to investigate candidate genes in these regions and to identify putative causative mutations that influence these traits. In addition, we tested the identified mutations for female fertility and growth traits. Results: Using a combination of bioinformatics and molecular assay technology, twelve non-synonymous SNPs in eleven genes were genotyped in a cattle population. Three and nine SNPs explained more than 1% of the additive genetic variance for percentage of normal sperm and scrotal circumference, respectively. The SNPs that had a major influence in percentage of normal sperm were mapped to LOC100138021 and TAF7L genes; and in TEX11 and AR genes for scrotal circumference. One SNP in TEX11 was explained ~13% of the additive genetic variance for scrotal circumference at 12 months. The tested SNP were also associated with weight measurements, but not with female fertility traits. -

Emi1 (FBXO5) (Center) Rabbit Polyclonal Antibody Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for AP51634PU-N Emi1 (FBXO5) (Center) Rabbit Polyclonal Antibody Product data: Product Type: Primary Antibodies Applications: WB Recommended Dilution: ELISA: 1/1000. Western Blot: 1/100-1/500. Reactivity: Human Host: Rabbit Isotype: Ig Clonality: Polyclonal Immunogen: KLH conjugated synthetic peptide between 191-221 amino acids from the Central region of Human FBXO5. Specificity: This antibody recognizes Human FBXO5 (Center). Formulation: PBS containing 0.09% (W/V) Sodium Azide as preservative State: Aff - Purified State: Liquid purified Ig fraction Concentration: lot specific Purification: Affinity Chromatography on Protein A Conjugation: Unconjugated Storage: Store undiluted at 2-8°C for one month or (in aliquots) at -20°C for longer. Avoid repeated freezing and thawing. Stability: Shelf life: one year from despatch. Gene Name: Homo sapiens F-box protein 5 (FBXO5), transcript variant 2 Database Link: Entrez Gene 26271 Human Q9UKT4 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Emi1 (FBXO5) (Center) Rabbit Polyclonal Antibody – AP51634PU-N Background: This gene encodes a member of the F-box protein family which is characterized by an approximately 40 amino acid motif, the F-box. The F-box proteins constitute one of the four subunits of the ubiquitin protein ligase complex called SCFs (SKP1-cullin-F-box), which function in phosphorylation-dependent ubiquitination. -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body.