Chromatin Loops Associated with Active Genes and Heterochromatin Shape Rice Genome Architecture for Transcriptional Regulation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
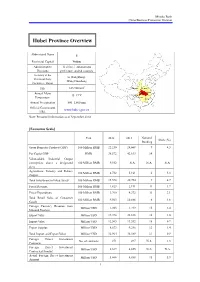
Hubei Province Overview
Mizuho Bank China Business Promotion Division Hubei Province Overview Abbreviated Name E Provincial Capital Wuhan Administrative 12 cities, 1 autonomous Divisions prefecture, and 64 counties Secretary of the Li Hongzhong; Provincial Party Wang Guosheng Committee; Mayor 2 Size 185,900 km Shaanxi Henan Annual Mean Hubei Anhui 15–17°C Chongqing Temperature Hunan Jiangxi Annual Precipitation 800–1,600 mm Official Government www.hubei.gov.cn URL Note: Personnel information as of September 2014 [Economic Scale] Unit 2012 2013 National Share (%) Ranking Gross Domestic Product (GDP) 100 Million RMB 22,250 24,668 9 4.3 Per Capita GDP RMB 38,572 42,613 14 - Value-added Industrial Output (enterprises above a designated 100 Million RMB 9,552 N.A. N.A. N.A. size) Agriculture, Forestry and Fishery 100 Million RMB 4,732 5,161 6 5.3 Output Total Investment in Fixed Assets 100 Million RMB 15,578 20,754 9 4.7 Fiscal Revenue 100 Million RMB 1,823 2,191 11 1.7 Fiscal Expenditure 100 Million RMB 3,760 4,372 11 3.1 Total Retail Sales of Consumer 100 Million RMB 9,563 10,886 6 4.6 Goods Foreign Currency Revenue from Million USD 1,203 1,219 15 2.4 Inbound Tourism Export Value Million USD 19,398 22,838 16 1.0 Import Value Million USD 12,565 13,552 18 0.7 Export Surplus Million USD 6,833 9,286 12 1.4 Total Import and Export Value Million USD 31,964 36,389 17 0.9 Foreign Direct Investment No. -

Characteristics of Metro Networks and Methodology for Their Evaluation
22 TRANSPORTATION RESEARCH RECORD 1162 Characteristics of Metro Networks and Methodology for Their Evaluation ANTONIO Musso AND VuKAN R. VucH1c PURPOSE, ORGANIZATION, AND SCOPE Presented In this paper are the results of research on metro (rapid transit) networks, focusing on their geometric charac teristics. The object Is to define the most important measures, Presented in this paper is a systematic set of quantitative Indicators, and characteristics of geometric forms that can elements that defines the network characteristics of metro sys Improve the present predominantly empirical methods used in tems that can be used for their description, evaluation, and metro network planning and analysis. Several measures of comparative analysis. Examples of such evaluations include metro network size and rorm, including length, number or planning of new or analysis of existing networks, their com lines, and stations, which express the extensiveness of the sys parison with networks of other cities, and comparison of alter tem, are selected; they are also needed for derivations of various indicators. A number of selected indicators are then native network extensions. presented. These represent the most effective tool for network The quantitative elements are grouped into five general cate comparison because most of them are Independent of network gories, as follows: size. Several Indicators relating metro network to the city size and population express the degree of adequacy of the network 1. Measures of network size and form, to meet the city's needs. Based on experiences from a number of metro systems, characteristics of different types of lines 2. Indicators of network topology, (radial, diametrical, circumferential, and other) are defined. -

Use Style: Paper Title
2019 4th International Conference on Education and Social Development (ICESD 2019) ISBN: 978-1-60595-621-3 Wuhan Subway Economic Development in the Perspective of "Innovation, Coordination, Green, Open, Sharing" Concept 1,a 2,b, Fang WANG and Xiao-sheng LEI * 1Hubei University of Traditional Chinese Medicine, Wuhan, Hubei, China 2Hubei University of Traditional Chinese Medicine, Wuhan, Hubei, China [email protected], [email protected] *Corresponding author Keywords: Development Concept, Wuhan, Subway Economy. Abstract. Since the 18th congress of the communist party of China, the "innovation, coordination, green, open and shared" development concept has been a profound change in the overall situation of China's development. In this context, the Wuhan Metro has developed rapidly, This paper analyzes the current situation, characteristics and influence of the development of subway economy in Wuhan from the perspective of development concept, and puts forward some suggestions for the future development of subway economy. Introduction For a long time, China has faced problems such as uneven, inadequate, uncoordinated and unsustainable development, and it has already faced the "ceiling dilemma". In recent years, China is facing the downward pressure of the economy. In order to change the development mode and solve the development problems, the 18th National Congress put forward five development concepts of “innovation, coordination, green, openness and sharing”, which have important guidance for the future development of social economy. Xiong'an New District is an innovative, coordinated, green ecological, open and co-constructed shared future urban model. Wuhan, following the glory of history, is becoming a reactivated city, and it must follow this development philosophy. -

Force Estimation Based on Semg Using Wavelet Analysis and Neural Network
Force Estimation Based on sEMG using Wavelet Analysis and Neural Network line 1: 1st Du Jiang line 1: 2ndGongfa Li line 1: 3rd Guozhang Jiang line 1: 4th Disi Chen line 2: Key Laboratory of line 2: Key Laboratory of line 2: Hubei Key Laboratory of line 2: School of Computing, Metallurgical Equipment and Metallurgical Equipment and Mechanical Transmission and University of Portsmouth Control Technology, Ministry of Control Technology, Ministry of Manufacturing Engineering, line3: Portsmouth, UK Education Wuhan University of Education Wuhan University of Wuhan University of Science and line 4: [email protected] Science and Technology Science and Technology Technology line 3: Hubei Key Laboratory of line 3: Research Center of line 3: 3D Printing and Mechanical Transmission and Biologic Manipulator and Intelligent Manufacturing line 1: 5th Zhaojie Ju Manufacturing Engineering, Intelligent Measurement and Engineering Institute, Wuhan line 2: School of Computing, Wuhan University of Science and Control, Wuhan University of University of Science and University of Portsmouth Technology Science and Technology Technology line 3: Portsmouth, UK line 4: Wuhan, China line 4: Wuhan, China line 4: Wuhan, China line 4: [email protected] line 5: [email protected] line 5: [email protected] line 5: [email protected] Abstract—In order to meet the needs of sEMG signal control gesture recognition and human-computer interaction[9]. The in human-computer interaction, an estimation of grip force based main work of sEMG-based human hand motion pattern on wavelet analysis and neural network is proposed. Firstly, the recognition is to study the action pattern of identifying the acquisition of EMG signals and the extraction methods of hand from the surface EMG signal, that is, the feature traditional features are described based on the introduction extraction and motion pattern recognition of sEMG[10], [11]. -

Ozone and Fine Particle in the Western Yangtze River Delta: an Overview Of
Atmos. Chem. Phys. Discuss., 13, C673–C680, 2013 Atmospheric www.atmos-chem-phys-discuss.net/13/C673/2013/ Chemistry © Author(s) 2013. This work is distributed under and Physics the Creative Commons Attribute 3.0 License. Discussions Interactive comment on “Ozone and fine particle in the western Yangtze River Delta: an overview of 1-yr data at the SORPES station” by A. J. Ding et al. Anonymous Referee #1 Received and published: 19 March 2013 General comments: The manuscript shows a 1 year data series of trace gases and PM2.5 observations in the recently set up SORPES station in East China. The site is under influence of urban and biomass burning emissions, being an interesting location to study the interactions between surface emissions and pollution transport. The dataset is of good quality and the data analysis consistent. I recommend publication in ACP after minor revisions, according to the following comments. Specific comments: 1. Section 2.1: You did not mention the construction works in the campus (Herman et C673 al., 2013). This could be a significant source or aerosols. Please discuss the potential impact on your results. 2. Page 2840, lines 11-13: It would be great to give the reader an idea of spatial distances, either by adding a length scale to Figure 1c, or by mentioning distances in the text. 3. Page 2840, line 19: what was the measurement height? 40 m above ground level? Or 40 m above the surrounding landscape? 4. Section 2.1: How about meteorological measurements? What equipment was used, and which variables were measured? 5. -

High-Resolution (0.05°X0.05°) Nox Emissions in the Yangtze River Delta Inferred from OMI”
Review “High-resolution (0.05°x0.05°) NOx emissions in the Yangtze River delta inferred from OMI” The manuscript entitled ‘High-resolution (0.05◦×0.05◦) NOx emissions in the Yangtze River Delta inferred from OMI’ presents an inventive method to estimate NOx emissions on a high resolution from satellite observations. The topic is interesting and important. Important aspect is also the error analysis that has been described in detail. However, before the paper gets published, the manuscript can be improved a lot by adding more discussion of the results including the relation to other existing inventories, and the description of method should be rephrased in a clearer way. General remarks This inventory is presented as the only high resolution inventory for the YRD region, but in the MarcoPolo-Panda project, a high resolution emission inventory of 0.01 degree resolution has been developed for this region (see http://www.marcopolo-panda.eu/products/toolbox/emission-data/) Since the affiliations of the authors were also participating in the MarcoPolo-Panda project it is surprising that this inventory is not mentioned or used in their comparisons. Also Zhao et al. (2015) present a city-scale emission inventory with the resolution of 3kmx3km in Nanjing, in the Yangtze River Delta.: Zhao, Y., Qiu, L. P., Xu, R. Y., Xie, F. J., Zhang, Q., Yu, Y. Y., Nielsen, C. P., Qin, H. X., Wang, H. K., Wu, X. C., Li, W. Q., and Zhang, J.: Advantages of a city-scale emission inventory for urban air quality research and policy: the case of Nanjing, a typical industrial city in the Yangtze River Delta, China, Atmos. -

Associating the COVID-19 Severity with Urban Factors: a Case Study of Wuhan
Associating the COVID-19 Severity with Urban Factors: A Case Study of Wuhan Xin Li School of Architecture and Civil Engineering, Xiamen University Lin Zhou School of Urban Design, Wuhan University Tao Jia ( [email protected] ) Wuhan University Ran Peng School of Civil Engineering and Architecture, Wuhan Institute of Technology Xiongwu Fu Wuhan Landuse and Urban Spatial Planning Research Center Yuliang Zou School of Public Health, Wuhan University Research Keywords: COVID-19, Weibo, epidemic pattern, spatial analysis, urban factors Posted Date: June 18th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-34863/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/28 Abstract Wuhan encountered a serious attack in the rst round of the COVID-19 pandemic which has resulted in serious worldwide consequences politically and economically. Based on the Weibo help data, we inferred the spatial distribution pattern of the epidemic situation and its impacts. Seven urban factors, i.e. urban growth, general hospital, commercial facilities, subway station, landuse mixture, aging ratio, and road density, were selected for validation with the ordinary linear model, and the former six presented globally signicant association with the epidemic severity; thereafter, the geographically weighted regression model was further adopted for local test to identify their unevenly distributed effects in urban space. Among the six, the place where general hospitals exert effects on epidemic situation highly is associated with their distribution and density; commercial facilities appear the most prevalently distributed factor over the city; newly developed residential quarters with high-rise buildings face greater risks, mainly distributed around the waterfront area of Hanyang and Wuchang; the inuence of subway stations concentrates at the adjacency place where the three towns meet and near-terminal locations; aging ratio dominantly affects the hinterland of Hankou in a broader extent than other areas in the city. -

Assessing the Impacts of High Speed Rail Development in China's
Prime Archives in Transportation and Logistics Book Chapter Assessing the Impacts of High Speed Rail Development in China’s Yangtze River Delta Megaregion Xueming Chen* L. Douglas Wilder School of Government and Public Affairs, Virginia Commonwealth University, USA *Corresponding Author: Xueming Chen, L. Douglas Wilder School of Government and Public Affairs, Virginia Commonwealth University, Richmond, United States Published February 10, 2020 This Book Chapter is a republication of an article published by Xueming Chen at Journal of Transportation Technologies in January 2013. (X. Chen, "Assessing the Impacts of High Speed Rail Development in China’s Yangtze River Delta Megaregion," Journal of Transportation Technologies, Vol. 3 No. 2, 2013, pp. 113-122. doi: 10.4236/jtts.2013.32011.) How to cite this book chapter: Xueming Chen. Assessing the Impacts of High Speed Rail Development in China’s Yangtze River Delta Megaregion. In: Prime Archives in Transportation and Logistics. Hyderabad, India: Vide Leaf. 2020. © The Author(s) 2020. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License(http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Acknowledgements: This author greatly appreciates the data support provided by Professor Haixiao Pan, Department of Urban Planning, Tongji University, China. The potential remaining errors of this paper are mine. 75 www.videleaf.com Prime Archives in Transportation and Logistics Abstract This paper assesses the impacts of high speed rail (HSR) development in the Yangtze River Delta (YRD) Megaregion, China. After giving an introduction and conducting a literature review, the paper proposes a pole-axis-network system (PANS) model guiding the entire study. -

Empirical Validation of Network Learning with Taxi GPS Data from Wuhan, China
IEEE ITS MAGAZINE, VOL. XX, NO. XX, AUGUST 2020 1 Empirical Validation of Network Learning with Taxi GPS Data from Wuhan, China Susan Jia Xu, Qian Xie, Joseph Y. J. Chow, and Xintao Liu have been proposed in the literature: inverse shortest path [9]; Abstract—In prior research, a statistically cheap method was inverse linear programs for an assortment of transportation developed to monitor transportation network performance by problems [10]; link capacities in minimum cost flow problems using only a few groups of agents without having to forecast the [11]; inverse vehicle routing problems [12;13]; general inverse population flows. The current study validates this “multi-agent variational inequalities for equilibrium models [14]; and route inverse optimization” method using taxi GPS trajectories data from the city of Wuhan, China. Using a controlled 2062-link choice [15]. network environment and different GPS data processing Despite the growing literature, inverse transportation algorithms, an online monitoring environment is simulated using problems are designed to take a system level model and the real data over a 4-hour period. Results show that using only estimate parameters of that model from sample data. This is samples from one OD pair, the multi-agent inverse optimization problematic because congested systems require estimation of method can learn network parameters such that forecasted travel times have a 0.23 correlation with the observed travel times. By population attributes like flow in order to quantify congestion increasing to monitoring from just two OD pairs, the correlation effect parameters because the more congested the system the improves further to 0.56. -

Wuhan Urban Rail Transit Line 2 Project
Why do you think this project should receive an award? How does it demonstrate: innovation, quality, and professional excellence transparency and integrity in the management and project implementation sustainability and respect for the environment Wuhan a richly historied city, is divided by Yangtze River the third longest river in the world. The city becomes prosperous due to the river but also has very inconvenient traffic conditions arising from the obstruction of the river. Wuhan Urban Rail Transit Line 2 Project is built for the purpose of solving the traffic problem crossing the Yangtze River, with a total length of 27.735 km and designed with 21 stations in total. It is the first fully-underground rail transit line in Wuhan City and the first subway line crossing Yangtze River in China, and furthermore, a key rail transit line with the maximum passenger flow as predicted in the URT network. This line has connected several important nodes in Wuhan City and is hailed as a golden URT line in Wuhan City. The study work of this project was commenced from 1996 and it was opened to traffic for trial operation on December 28, 2012. It primarily solves the river-crossing problem in Wuhan City, alleviates the traffic pressure on Wuhan Yangtze River Bridge and The Second Wuhan Yangtze River Bridge and assumes 50% of river-crossing passenger flow realized by public transit system. I. Arduous and Complicated Work, Leading Innovation Achievements Wuhan URT Line 2 Project is a huge systematic endeavor that involves with more than 30 specialties. Its significant functional orientation and particular engineering geological & hydrological conditions present a huge challenge to the engineering construction in the history of subway construction all over the world. -
The Implementation of Financing Strategies in Urban Rapid Transit Infrastructure: How Could Chinese Cities Do Better?
The Implementation of Financing Strategies in Urban Rapid Transit Infrastructure: How Could Chinese Cities Do Better? A Thesis Presented to the Faculty of Architecture and Planning COLUMBIA UNIVERSITY In Partial Fulfillment of the Requirements for the Degree Master of Science in Urban Planning by Yifei Ma May 2016 Acknowledgements This thesis attempts study the stories of many Chinese mainland cities, which have been developing transit infrastructure at an incredible speed. Each city has its own history and blueprint. Hopefully, this thesis could portray the decision-making process of how the cities made their own adaptions in learning advanced experience, especially in the contract design and the role of government, and the implementation process. First and foremost, I want to thank my thesis advisor Dr. Elliott Sclar for his insights and suggestions. I had a difficult time in streamlining the research and picturing the story. It was Dr. Sclar who utilized his expertise and experience in China to help me figure out the direction and offered valuable suggestions to improve this thesis. I extend a special thanks to Dr. David King, who graciously agreed to take time from his busy schedule to be my second reader. Finally, I want to express my deep gratitude to my parents, whose constant love, support, and encouragement are instrumental in having my studies in the Urban Planning Program at GSAPP and completing the thesis. I dedicate this thesis to my mother and father. Abstract Urban rapid transit infrastructure have been expanding at an exploding speed in Mainland China. Government subsidy used to be the sole fiscal support to the transit development for a long time. -

Detailed Strategies for the Global Finals of the 28Th World Memory
Detailed strategies for the global finals of the 28th World Memory Championships The 28th World Memory Championships 2019 global finals look forward to your arrival The clarion call for mental assembly has sounded! The global finals of the 28th World Memory Championships in 2019 are about to begin! From the WMC of city level,national Level to the international level,spanning three months,this is a journey of exploring the limits of the memory, this is a journey of chasing the dream of the world memory masters, this is a journey of winning glory for China's mental sports Olympiad of mind sports makes the world intelligent! Promotional video for the global finals of the 28th World Memory Championships Wuhan city image promotion video Big City Rise (English version) This is the annual memory event, it is the highest glory of the global memory to compete here. Welcome the world's memory power experts to jointly create history and explore the limits of human memory abilities. Event Introduction Guiding unit: World Memory Sport Council Asia Pacific World Memory Sports Council Host unit: Administrative committee of Wuhan East Lake high-tech Development Zone Organizer: Wuhan Broadcasting and Television Station Wuhan Haijing Education TechNo.logy co.ltd World Memory Championships (WMC) was initiated in 1991 by professor Tony Buzan, the father of World Memory, inventor of mind mapping, and Sir Raymond keene, the British OBE Medal winner. It is a globally high-level brainpower competition organized by the World Memory Sports Council and is hoNo.red as the Olympics of brain sports 1.