DNA Copy Number Aberrations Associated with Aneuploidy and Chromosomal Instability in Breast Cancers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Produktinformation
Produktinformation Diagnostik & molekulare Diagnostik Laborgeräte & Service Zellkultur & Verbrauchsmaterial Forschungsprodukte & Biochemikalien Weitere Information auf den folgenden Seiten! See the following pages for more information! Lieferung & Zahlungsart Lieferung: frei Haus Bestellung auf Rechnung SZABO-SCANDIC Lieferung: € 10,- HandelsgmbH & Co KG Erstbestellung Vorauskassa Quellenstraße 110, A-1100 Wien T. +43(0)1 489 3961-0 Zuschläge F. +43(0)1 489 3961-7 [email protected] • Mindermengenzuschlag www.szabo-scandic.com • Trockeneiszuschlag • Gefahrgutzuschlag linkedin.com/company/szaboscandic • Expressversand facebook.com/szaboscandic SANTA CRUZ BIOTECHNOLOGY, INC. OR2M5 siRNA (h): sc-88564 BACKGROUND STORAGE AND RESUSPENSION Olfactory receptors are G protein-coupled receptor proteins that localize to the Store lyophilized siRNA duplex at -20° C with desiccant. Stable for at least cilia of olfactory sensory neurons where they display affinity for and bind to one year from the date of shipment. Once resuspended, store at -20° C, a variety of odor molecules. The genes encoding olfactory receptors comprise avoid contact with RNAses and repeated freeze thaw cycles. the largest family in the human genome. The binding of olfactory receptor Resuspend lyophilized siRNA duplex in 330 µl of the RNAse-free water proteins to odor molecules triggers a signal transduction cascade that leads provided. Resuspension of the siRNA duplex in 330 µl of RNAse-free water to the production of cAMP via an olfactory-enriched adenylate cyclase. This makes a 10 µM solution in a 10 µM Tris-HCl, pH 8.0, 20 mM NaCl, 1 mM event ultimately leads to transmission of action potentials to the brain and EDTA buffered solution. the subsequent perception of smell. -

The Genetic Basis of Dupuytren's Disease Gloria Sue Yale School of Medicine, [email protected]
Yale University EliScholar – A Digital Platform for Scholarly Publishing at Yale Yale Medicine Thesis Digital Library School of Medicine January 2014 The Genetic Basis Of Dupuytren's Disease Gloria Sue Yale School of Medicine, [email protected] Follow this and additional works at: http://elischolar.library.yale.edu/ymtdl Recommended Citation Sue, Gloria, "The Genetic Basis Of Dupuytren's Disease" (2014). Yale Medicine Thesis Digital Library. 1926. http://elischolar.library.yale.edu/ymtdl/1926 This Open Access Thesis is brought to you for free and open access by the School of Medicine at EliScholar – A Digital Platform for Scholarly Publishing at Yale. It has been accepted for inclusion in Yale Medicine Thesis Digital Library by an authorized administrator of EliScholar – A Digital Platform for Scholarly Publishing at Yale. For more information, please contact [email protected]. The Genetic Basis of Dupuytren’s Disease A Thesis Submitted to the Yale University School of Medicine In Partial Fulfillment of the Requirements for the Degree of Doctor of Medicine by Gloria R. Sue 2014 THE GENETIC BASIS OF DUPUYTREN’S DISEASE. Gloria R. Sue, Deepak Narayan. Section of Plastic and Reconstructive Surgery, Department of Surgery, Yale University School of Medicine, New Haven, CT. Dupuytren’s disease is a common heritable connective tissue disorder of poorly understood etiology. It is thought that oxidative stress pathways may play a critical role in the development of Dupuytren’s disease, given the various disease associations that have been observed. We sought to sequence the mitochondrial and nuclear genomes of patients affected with Dupuytren’s disease using next-generation sequencing technology to potentially identify genes of potential pathogenetic interest. -

Identification of Candidate Biomarkers and Pathways Associated with Type 1 Diabetes Mellitus Using Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.08.447531; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Identification of candidate biomarkers and pathways associated with type 1 diabetes mellitus using bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.06.08.447531; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Type 1 diabetes mellitus (T1DM) is a metabolic disorder for which the underlying molecular mechanisms remain largely unclear. This investigation aimed to elucidate essential candidate genes and pathways in T1DM by integrated bioinformatics analysis. In this study, differentially expressed genes (DEGs) were analyzed using DESeq2 of R package from GSE162689 of the Gene Expression Omnibus (GEO). Gene ontology (GO) enrichment analysis, REACTOME pathway enrichment analysis, and construction and analysis of protein-protein interaction (PPI) network, modules, miRNA-hub gene regulatory network and TF-hub gene regulatory network, and validation of hub genes were then performed. A total of 952 DEGs (477 up regulated and 475 down regulated genes) were identified in T1DM. GO and REACTOME enrichment result results showed that DEGs mainly enriched in multicellular organism development, detection of stimulus, diseases of signal transduction by growth factor receptors and second messengers, and olfactory signaling pathway. -

WO 2019/068007 Al Figure 2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/068007 Al 04 April 2019 (04.04.2019) W 1P O PCT (51) International Patent Classification: (72) Inventors; and C12N 15/10 (2006.01) C07K 16/28 (2006.01) (71) Applicants: GROSS, Gideon [EVIL]; IE-1-5 Address C12N 5/10 (2006.0 1) C12Q 1/6809 (20 18.0 1) M.P. Korazim, 1292200 Moshav Almagor (IL). GIBSON, C07K 14/705 (2006.01) A61P 35/00 (2006.01) Will [US/US]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., C07K 14/725 (2006.01) P.O. Box 4044, 7403635 Ness Ziona (TL). DAHARY, Dvir [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (21) International Application Number: Box 4044, 7403635 Ness Ziona (IL). BEIMAN, Merav PCT/US2018/053583 [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (22) International Filing Date: Box 4044, 7403635 Ness Ziona (E.). 28 September 2018 (28.09.2018) (74) Agent: MACDOUGALL, Christina, A. et al; Morgan, (25) Filing Language: English Lewis & Bockius LLP, One Market, Spear Tower, SanFran- cisco, CA 94105 (US). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of national protection available): AE, AG, AL, AM, 62/564,454 28 September 2017 (28.09.2017) US AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, 62/649,429 28 March 2018 (28.03.2018) US CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, (71) Applicant: IMMP ACT-BIO LTD. -
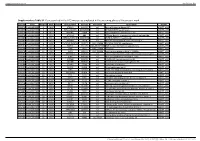
Supplementary Table S1. Genes Printed in the HC5 Microarray Employed in the Screening Phase of the Present Work
Supplementary material Ann Rheum Dis Supplementary Table S1. Genes printed in the HC5 microarray employed in the screening phase of the present work. CloneID Plate Position Well Length GeneSymbol GeneID Accession Description Vector 692672 HsxXG013989 2 B01 STK32A 202374 null serine/threonine kinase 32A pANT7_cGST 692675 HsxXG013989 3 C01 RPS10-NUDT3 100529239 null RPS10-NUDT3 readthrough pANT7_cGST 692678 HsxXG013989 4 D01 SPATA6L 55064 null spermatogenesis associated 6-like pANT7_cGST 692679 HsxXG013989 5 E01 ATP1A4 480 null ATPase, Na+/K+ transporting, alpha 4 polypeptide pANT7_cGST 692689 HsxXG013989 6 F01 ZNF816-ZNF321P 100529240 null ZNF816-ZNF321P readthrough pANT7_cGST 692691 HsxXG013989 7 G01 NKAIN1 79570 null Na+/K+ transporting ATPase interacting 1 pANT7_cGST 693155 HsxXG013989 8 H01 TNFSF12-TNFSF13 407977 NM_172089 TNFSF12-TNFSF13 readthrough pANT7_cGST 693161 HsxXG013989 9 A02 RAB12 201475 NM_001025300 RAB12, member RAS oncogene family pANT7_cGST 693169 HsxXG013989 10 B02 SYN1 6853 NM_133499 synapsin I pANT7_cGST 693176 HsxXG013989 11 C02 GJD3 125111 NM_152219 gap junction protein, delta 3, 31.9kDa pANT7_cGST 693181 HsxXG013989 12 D02 CHCHD10 400916 null coiled-coil-helix-coiled-coil-helix domain containing 10 pANT7_cGST 693184 HsxXG013989 13 E02 IDNK 414328 null idnK, gluconokinase homolog (E. coli) pANT7_cGST 693187 HsxXG013989 14 F02 LYPD6B 130576 null LY6/PLAUR domain containing 6B pANT7_cGST 693189 HsxXG013989 15 G02 C8orf86 389649 null chromosome 8 open reading frame 86 pANT7_cGST 693194 HsxXG013989 16 H02 CENPQ 55166 -

Chromophobe Renal Cell Carcinoma with and Without Sarcomatoid Change: a Clinicopathological, Comparative Genomic Hybridization, and Whole-Exome Sequencing Study
Am J Transl Res 2015;7(11):2482-2499 www.ajtr.org /ISSN:1943-8141/AJTR0014993 Original Article Chromophobe renal cell carcinoma with and without sarcomatoid change: a clinicopathological, comparative genomic hybridization, and whole-exome sequencing study Yuan Ren1*, Kunpeng Liu1*, Xueling Kang3, Lijuan Pang1, Yan Qi1, Zhenyan Hu1, Wei Jia1, Haijun Zhang1, Li Li1, Jianming Hu1, Weihua Liang1, Jin Zhao1, Hong Zou1*, Xianglin Yuan2, Feng Li1* 1Department of Pathology, School of Medicine, First Affiliated Hospital, Shihezi University, Key Laboratory of Xinjiang Endemic and Ethnic Diseases, Ministry of Education of China, Shihezi, China; 2Tongji Hospital Cancer Center, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China; 3Department of Pathology, Shanghai General Hospital, Shanghai, China. *Equal contributors. Received August 24, 2015; Accepted October 13, 2015; Epub November 15, 2015; Published November 30, 2015 Abstract: Chromophobe renal cell carcinomas (CRCC) with and without sarcomatoid change have different out- comes; however, fewstudies have compared their genetic profiles. Therefore, we identified the genomic alteration- sin CRCC common type (CRCC C) (n=8) and CRCC with sarcomatoid change (CRCC S) (n=4) using comparative genomic hybridization (CGH) and whole-exome sequencing. The CGH profiles showed that the CRCC C group had more chromosomal losses (72 vs. 18) but fewer chromosomal gains (23 vs. 57) than the CRCC S group. Losses of chromosomes 1p, 8p21-23, 10p16-20, 10p12-ter, 13p20-30, and 17p13 and gains of chromosomes 1q11, 1q21-23, 1p13-15, 2p23-24, and 3p21-ter differed between the groups. Whole-exome sequencing showed that the mutational status of 270 genes differed between CRCC (n=12) and normal renal tissues (n=18). -
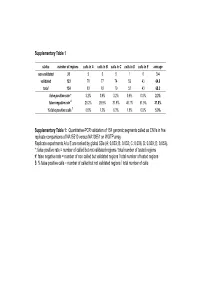
Quantitative-PCR Validation of 154 Genomic Segments Called As Cnvs in Five Replicat
Supplementary Table 1 status number of regions calls in A calls in B calls in C calls in D calls in E average non validated 31 5 6 5 1 0 3.4 validated 123 78 77 74 52 43 64.8 total 154 83 83 79 53 43 68.2 false positive rate * 3.2% 3.9% 3.2% 0.6% 0.0% 2.2% false negative rate # 29.2% 29.9% 31.8% 46.1% 51.9% 37.8% % false positive calls $ 6.0% 7.2% 6.3% 1.9% 0.0% 5.0% Supplementary Table 1: Quantitative-PCR validation of 154 genomic segments called as CNVs in five replicate comparisons of NA15510 versus NA10851 on WGTP array Replicate experiments A to E are ranked by global SDe (A: 0.033; B: 0.033; C: 0.036; D: 0.039; E: 0.053). *: false positive rate = number of called but not validated regions / total number of tested regions #: false negative rate = number of non called but validated regions / total number of tested regions $: % false positive calls = number of called but not validated regions / total number of calls False positive estimates for 500K EA CNV calls Total Rep1 Rep2 Rep3 Avg (unique) Validated 33 28 32 31 38 Not validated 2 2 2 2 5 Total 35 30 34 33 43 % False positive 5.71% 6.67% 5.88% 6.09% - % False negative 13.16% 26.32% 15.79% 18.42% - Supplementary Table 2A : Quantitative PCR validation of 43 unique CNV regions called as CNVs in three replicate comparisons of NA15510 versus NA10851 using the 500K EA array. -

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -

Table S4. RAE Analysis of Dedifferentiated Liposarcoma
Table S4. RAE analysis of dedifferentiated liposarcoma Model Chromosome Region start Region end Size q value freqX0* # genes genes Amp 1 57809872 60413476 2603605 0.00026 34.6 10 DAB1,RPS26P15,OMA1,TACSTD2,MYSM1,JUN,FGGY,HOOK1,CYP2J2,C1orf87 Amp 1 158619146 158696968 77823 0.053 25 1 VANGL2 Amp 1 158883523 158922841 39319 0.081 23.1 2 SLAMF1,CD48 Amp 1 162042586 162118557 75972 0.072 25 0 [Nearest:NUF2] Amp 1 162272460 162767627 495168 0.017 26.9 0 [Nearest:PBX1] Amp 1 165486554 165532374 45821 0.057 25 1 POU2F1 Amp 1 167138282 167483267 344986 0.024 26.9 2 ATP1B1,NME7 Amp 1 167612872 167708844 95973 0.041 25 3 BLZF1,C1orf114,SLC19A2 Amp 1 167728199 167808161 79963 0.076 21.2 1 F5 Amp 1 168436370 169233893 797524 0.018 26.9 3 GORAB,PRRX1,C1orf129 Amp 1 169462231 170768440 1306210 1.3E-06 38.5 10 FMO1,FMO4,TOP1P1,BAT2D1,MYOC,VAMP4,METTL13,DNM3,C1orf105,PIGC Amp 1 171026247 171291427 265181 0.015 26.9 1 TNFSF18 Del 1 201860394 202299299 438906 0.0047 25 6 ATP2B4,SNORA77,LAX1,ZC3H11A,SNRPE,C1orf157 Del 1 210909187 212021116 1111930 0.017 19.2 8 BATF3,NSL1,TATDN3,C1orf227,FLVCR1,VASH2,ANGEL2,RPS6KC1 Del 1 215937857 216049214 111358 0.079 23.1 1 SPATA17 Del 1 218237257 218367476 130220 0.0063 26.9 3 EPRS,BPNT1,IARS2 Del 1 222100886 222727238 626353 5.2E-05 32.7 5 FBXO28,DEGS1,NVL,CNIH4,WDR26 Del 1 223166548 224519805 1353258 0.0063 26.9 15 DNAH14,LBR,ENAH,SRP9,EPHX1,TMEM63A,LEFTY1,PYCR2,LEFTY2,C1orf55,H3F3A,LOC440926 ,ACBD3,MIXL1,LIN9 Del 1 225283136 225374166 91031 0.054 23.1 1 CDC42BPA Del 1 227278990 229012661 1733672 0.091 21.2 13 RAB4A,SPHAR,C1orf96,ACTA1,NUP133,ABCB10,TAF5L,URB2,GALNT2,PGBD5,COG2,AGT,CAP -

1 Copper-Mediated Thiol Potentiation and Mutagenesis-Guided Modeling Suggest a Highly Conserved Copper Binding Motif in Human OR
Copper-mediated thiol potentiation and mutagenesis-guided modeling suggest a highly conserved copper binding motif in human OR2M3 Franziska Haag1, Lucky Ahmed2, Krystle Reiss2, Eric Block3, Victor S. Batista2 and Dietmar Krautwurst1 1Leibniz-Institute for Food Systems Biology at the Technical University of Munich, Lise-Meitner-Str. 34, D-85354 Freising, Germany 2Department of Chemistry, Yale University, New Haven, CT 06520, United States 3Department of Chemistry, University at Albany, State University of New York, Albany, NY 12222, United States Contents: Table S1: Oligonucleotides for molecular cloning of odorant receptors investigated. ........................... 2 Table S2: Oligonucleotides for Homo sapiens OR2M3 site-directed mutagenesis. ................................ 2 Table S3: Oligonucleotides for Homo sapiens OR2W1 site-directed mutagenesis. ................................ 4 Table S4: Vector internal oligonucleotides for pi2-dk (39aa rho-tag). .................................................... 4 Table S5: NCBI reference sequences of olfactory receptor genes investigated. ..................................... 5 Table S6: EC50 values and relative amplitudes for 3-mercapto-2-methylpentan-1-ol on OR2M3 wild type in the absence and presence of different heavy metals. ................................................................ 7 Table S7: EC50 values and relative amplitudes for OR2M3 wild type in the absence and presence of different concentrations of Cu2+. ........................................................................................................... -

AKT-Mtor Signaling in Human Acute Myeloid Leukemia Cells and Its Association with Adverse Prognosis
Cancers 2018, 10, 332 S1 of S35 Supplementary Materials: Clonal Heterogeneity Reflected by PI3K- AKT-mTOR Signaling in Human Acute Myeloid Leukemia Cells and its Association with Adverse Prognosis Ina Nepstad, Kimberley Joanne Hatfield, Tor Henrik Anderson Tvedt, Håkon Reikvam and Øystein Bruserud Figure S1. Detection of clonal heterogeneity for 49 acute myeloid leukemia (AML) patients; the results from representative flow cytometric analyses of phosphatidylinositol-3-kinase-Akt-mechanistic target of rapamycin (PI3K-Akt-mTOR) activation. For each patient clonal heterogeneity was detected by analysis Cancers 2018, 10, 332 S2 of S35 of at least one mediator in the PI3K-Akt-mTOR pathway. Patient ID is shown in the upper right corner of each histogram. The figure documents the detection of dual populations for all patients, showing the results from one representative flow cytometric analysis for each of these 49 patients. The Y-axis represents the amount of cells, and the X-axis represents the fluorescence intensity. The stippled line shows the negative/unstained controls. Figure S2. Cell preparation and gating strategy. Flow cytometry was used for examination of the constitutive expression of the mediators in the PI3K-Akt-mTOR pathway/network in primary AML cells. Cryopreserved cells were thawed and washed before suspension cultures were prepared as described in Materials and methods. Briefly, cryopreserved and thawed primary leukemic cells were incubated for 20 minutes in RPMI-1640 (Sigma-Aldrich) before being directly fixed in 1.5% paraformaldehyde (PFA) and permeabilized with 100% ice-cold methanol. The cells were thereafter rehydrated by adding 2 mL phosphate buffered saline (PBS), gently re-suspended and then centrifuged. -

Differing from Diseased Stage in Melanoma, the Olfactory Receptor Gene OR56A4 Is Expressed
Differing from diseased stage in melanoma, the olfactory receptor gene OR56A4 is expressed Bronte Morse, Kobi Decker, Khushbu P, James K., Rushabh P Abstract We have compared the global profiles of 100 tumors in Stage I, II and III with two independently released microarray datasets in order to understand their transcriptional behaviors accompanying a progression in breast cancer (1, 2). The olfactive receptor, family 56, subfamily A, member 4 OR56A4, was discovered to have been one of the genes with the most varied expression when comparing initial tumors in stage I, stage II, and stage III of breast cancer patients. In the stage III tumors, OR56A4 expression in comparison to the stage I tumors was lower. Keywords: cancer, tumor transcriptome analysis, breast cancer, comparative differential gene expression profiling, systems biology of breast cancer. INTRODUCTION In nearly 275,000 women in the United States, breast cancer was diagnosed last year (3). While for women with stage I breast cancer, 5-year survival rates are 99 percent, for women with stage IV breast cancer, the 5-year survival rate is 27 percent (3). We examined 100 main tumor transcriptomas using 2 independently published microarray datasets to investigate how the transcription behavior of primary tumors in women with breast cancer varies according on disease stage (1, 2). OR56A4 was one of the most differentially expressed genes for both data sets when comparing disease-based tumor transcriptomes. METHOD For this differential analysis of gene expression along with GEO2R, GSE109169(1) and GSE86166(2) data sets have been utilized. A total of n=25 tumors with n=6 stage I, N =11 stage II, and n=7 stage III tumors were created from early start patients with breast cancers utilizing Affymetrix Human Exon 1,0 ST Array.