WM White Geochemistry Chapter 7
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Experimental Partitioning of Rb, Cs, Sr, and Ba Between Alkali Feldspar and Peraluminous Melt
American Mineralogist, Volume 81, pages 719-734,1996 Experimental partitioning of Rb, Cs, Sr, and Ba between alkali feldspar and peraluminous melt JONATHAN IcENHOWER AND DAVID LoNDON School of Geology and Geophysics, University of Oklahoma, 100 East Boyd Street, SEC 810, Norman, Oklahoma 73019, U.S.A. ABSTRACT Hydrous partial melting experiments performed between 650 and 750 °C at 200 MPa (H20) on synthetic metapelite compositions (quartz + albite + muscovite + biotite min- eral mixtures) doped with Ba, Sr, Rb, and Cs yielded alkali feldspar crystals with a wide range of compositions in equilibrium at their rims with peraluminous melt. Measured partition coefficients for normally trace lithophile elements between feldspar and melt [D(M)FSP/gl,M = Ba, Sr, Rb, Cs] do not depend on either temperature or bulk composition of melt for the compositions studied. Values of D(Sr)FSP/glare between 10 and 14 and appear to be independent of the albite and orthoclase contents of the feldspar crystals. In contrast, values of D(Ba)FSP/gland D(Rb)FsP/glare strongly dependent on the orthoclase content of feldspar, relationships that can be expressed by the following linear equations: D(Ba)FSP/gl= 0.07 + 0.25(orthoclase) and D(Rb)FsP/gl= 0.03 + 0.0 1(orthoclase), where orthoclase is in mole percent. These equations reproduce the range of previously reported values for D(Ba) and D(Rb) determined on natural and synthetic samples. A single par- tition coefficient for Cs was also determined at D(CS)Fsp/gl= 0.13. These data can be used in conjunction with recently published partition coefficients for muscovite, biotite, and plagioclase feldspars (Blundy and Wood 1991; Icenhower and London 1995) to model quantitatively the trace element signatures of peraluminous mag- mas during anatexis and crystallization. -
Using Rare Earth Elements to Model Silicate Melting and Crystallization
Using Rare Earth Elements to Model Silicate Melting and Crystallization The Rare Earth Elements (REE) The lanthanide series is formed by filling of 4f orbitals, and its constituents are commonly termed the rare earth elements (REE). The common valence state is +3 for all REE over a wide range of oxygen fugacity; Ce+4 can occur in highly oxidized environments at the earth's surface, and Eu+2 in reducing environments in the crust and mantle. The rare earth elements are lithophile elements (low electronegativities lead to the formation of highly ionic bonds), which substitute into many silicates and phosphates. Ionic radii of the lanthanides decreases with increasing atomic number from La to Lu, the lanthanide contraction… Because of their high charge and large radii, the rare earth elements are usually relatively incompatible in silicate minerals. However, due to the lanthanide contraction, the heavier rare earths are smaller and thus can "fit" within some lattice sites (although there must be charge compensation for the 3+ valence), so incompatibility decreases with increasing Z. Class Discussion #1: What mineral lattice sites could host the heavy rare earth elements? What about Eu2+? REE diagrams If we plot the concentrations of the rare earth elements as a group on a diagram of abundance versus atomic number, we observe two cosmochemical phenomena: 1) the decrease in absolute abundance with increasing atomic number; 2) the “even-odd effect” in relative abundances (higher abundances of even atomic number elements). Because these cosmochemical effects are common to all terrestrial rocks, we would rather ignore them in order to accentuate more subtle variations in absolute and relative concentrations of REE between samples. -
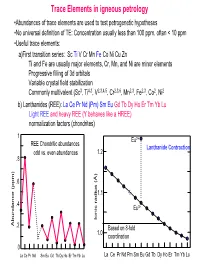
Trace Elements in Igneous Petrology
Trace Elements in igneous petrology •Abundances of trace elements are used to test petrogenetic hypotheses •No universal definition of TE: Concentration usually less than 100 ppm, often < 10 ppm •Useful trace elements: a)First transition series: Sc Ti V Cr Mn Fe Co Ni Cu Zn Ti and Fe are usually major elements, Cr, Mn, and Ni are minor elements Progressive filling of 3d orbitals Variable crystal field stabilization Commonly multivalent (Sc3, Ti4,3, V2,3,4,5, Cr2,3,6, Mn2,3, Fe2,3, Co2, Ni2 b) Lanthanides (REE): La Ce Pr Nd (Pm) Sm Eu Gd Tb Dy Ho Er Tm Yb Lu Light REE and heavy REE (Y behaves like a HREE) normalization factors (chondrites) 1 Eu2+ REE Chondritic abundances Lanthanide Contraction odd vs. even abundances 1.2 .8 .6 1.1 .4 Eu3+ Ionic radius (Å) Ionic radius Abundance (ppm) .2 Based on 8-fold 1.0 coordination 0 La Ce Pr Nd Sm Eu Gd Tb Dy Ho Er Tm Yb Lu La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu (c) Large Ion Lithophile Elements (LILE): may also be partitioned into fluid phase Alkalis: K Rb Cs (monovalent) Alkaline earths: Ba Sr (divalent) Actinides: U, Th, Ra, Pa (multiple valency) (d) High field strength elements (HFSE): small, highly-charged ions Zr, Hf (4 valent) Nb, Ta (4 and 5 valent) (e) Chalcophile elements: Cu, Zn, Pb, Ag, Hg, PGE, (Fe, Co, Ni) (f) Siderophile elements: Fe, Ni, Co, Ge, P, Ga, Au (PGE)… • Decoupled from major elements: lack of stoichiometric constraints (not strictly true) • Goldschmidt’s Rules • Generalities: Incompatible elements are elements that tend to be excluded from common minerals (olivines, pyroxenes, garnets, feldspars, oxides…) in equilibrium with a melt, i.e., they have low D values. -

Implications of Subduction Rehydration for Earth's Deep Water
Implications of Subduction Rehydration for Earth’s Deep Water Cycle Lars Rüpke Physics of Geological Processes, University of Oslo, Oslo, Norway, and SFB 574 Volatiles and Fluids in Subduction Zones, Kiel, Germany Jason Phipps Morgan Cornell University, Ithaca, New York, USA and SFB 574 Volatiles and Fluids in Subduction Zones, Kiel, Germany Jacqueline Eaby Dixon RSMAS/MGG, University of Miami, Miami, Florida, USA The “standard model” for the genesis of the oceans is that they are exhalations from Earth’s deep interior continually rinsed through surface rocks by the global hydrologic cycle. No general consensus exists, however, on the water distribution within the deeper mantle of the Earth. Recently Dixon et al. [2002] estimated water concentrations for some of the major mantle components and concluded that the most primitive (FOZO) are significantly wetter than the recycling associated EM or HIMU mantle components and the even drier depleted mantle source that melts to form MORB. These findings are in striking agreement with the results of numerical modeling of the global water cycle that are presented here. We find that the Dixon et al. [2002] results are consistent with a global water cycle model in which the oceans have formed by efficient outgassing of the mantle. Present-day depleted mantle will contain a small volume fraction of more primitive wet mantle in addition to drier recycling related enriched components. This scenario is consis- tent with the observation that hotspots with a FOZO-component in their source will make wetter basalts than hotspots whose mantle sources contain a larger fraction of EM and HIMU components. -

The Transition-Zone Water Filter Model for Global Material Circulation: Where Do We Stand?
The Transition-Zone Water Filter Model for Global Material Circulation: Where Do We Stand? Shun-ichiro Karato, David Bercovici, Garrett Leahy, Guillaume Richard and Zhicheng Jing Yale University, Department of Geology and Geophysics, New Haven, CT 06520 Materials circulation in Earth’s mantle will be modified if partial melting occurs in the transition zone. Melting in the transition zone is plausible if a significant amount of incompatible components is present in Earth’s mantle. We review the experimental data on melting and melt density and conclude that melting is likely under a broad range of conditions, although conditions for dense melt are more limited. Current geochemical models of Earth suggest the presence of relatively dense incompatible components such as K2O and we conclude that a dense melt is likely formed when the fraction of water is small. Models have been developed to understand the structure of a melt layer and the circulation of melt and vola- tiles. The model suggests a relatively thin melt-rich layer that can be entrained by downwelling current to maintain “steady-state” structure. If deep mantle melting occurs with a small melt fraction, highly incompatible elements including hydro- gen, helium and argon are sequestered without much effect on more compatible elements. This provides a natural explanation for many paradoxes including (i) the apparent discrepancy between whole mantle convection suggested from geophysical observations and the presence of long-lived large reservoirs suggested by geochemical observations, (ii) the helium/heat flow paradox and (iii) the argon paradox. Geophysical observations are reviewed including electrical conductivity and anomalies in seismic wave velocities to test the model and some future direc- tions to refine the model are discussed. -

Incompatible Element Ratios in Plate Tectonic and Stagnant Lid Planets Kent C. Condie1 and Charles K. Shearer 2. 1Department Of
47th Lunar! and Planetary Science Conference (2016) 1058.pdf Incompatible Element Ratios in Plate Tectonic and Stagnant Lid Planets Kent C. Condie1 and Charles K. Shearer2. 1Department of Earth and Environmental Science, New Mexico Tech, Socorro, New Mexico 87801, 2 Institute of Meteoritics, Department of Earth and Planetary Science, University of New Mexico, Albuquerque, New Mexico 87131, 2 NASA Johnson Space Center, Houston, TX 77058. Introduction: Because ratios of elements with group centered near Earth’s primitive mantle (PM) similar incompatibilities do not change much with composition (Fig. 2). This distribution is similar to degree of melting or fractional crystallization in the the distribution of most mare basalts from the Moon mantles of silicate planets, they are useful in sampled by the Apollo missions, except for the high- characterizing compositions of mantle sources of Ti basalts from Apollo 11 and 17 both of which are basalts that have not interacted with continental crust enriched in Nb. The primitive mantle grouping is [1,2]. Using Nb/Th and Zr/Nb ratios in young especially evident for Apollo 11, 12 and 15 basalts in oceanic basalts, three mantle domains can be which samples cluster tightly around primitive identified [3]: enriched (EM), depleted (DM), and mantle (PM) composition on Zr/Nb-Nb/Th, Th/Yb- hydrated mantle (HM) (Fig. 1). DM is characterized Nb/Yb and La/Sm-Nb/Th diagrams (Fig. 3). Very by high values of both ratios (Nb/Th > 8; Zr/Nb > 20) low-Ti green volcanic glasses (and some due to Th depletion relative to Nb, and to Nb intermediate-Ti yellow volcanic glasses) plot in a depletion relative to Zr. -

Durham Research Online
Durham Research Online Deposited in DRO: 14 March 2018 Version of attached le: Accepted Version Peer-review status of attached le: Peer-reviewed Citation for published item: Namur, Olivier and Humphreys, Madeleine C.S. (2018) 'Trace element constraints on the dierentiation and crystal mush solidication in the Skaergaard intrusion, Greenland.', Journal of petrology., 59 (3). pp. 387-418. Further information on publisher's website: https://doi.org/10.1093/petrology/egy032 Publisher's copyright statement: This is a pre-copyedited, author-produced version of an article accepted for publication in Journal Of Petrology following peer review. The version of record Namur, Olivier Humphreys, Madeleine C.S. (2018). Trace element constraints on the dierentiation and crystal mush solidication in the Skaergaard intrusion, Greenland. Journal of Petrology is available online at: https://doi.org/10.1093/petrology/egy032. Additional information: Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in DRO • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full DRO policy for further details. Durham University Library, Stockton Road, Durham DH1 3LY, United Kingdom Tel : +44 (0)191 334 3042 | Fax : +44 (0)191 334 2971 https://dro.dur.ac.uk Trace element constraints on the differentiation and crystal mush solidification in the Skaergaard intrusion, Greenland Olivier Namur1*, Madeleine C.S. -

WM White Geochemistry Chapter 7: Trace Elements
W. M. White Geochemistry Chapter 7: Trace Elements Chapter 7: Trace Elements in Igneous Processes 7.1 INTRODUCTION n this chapter we will consider the behavior of trace elements, particularly in magmas, and in- troduce methods to model this behavior. Though trace elements, by definition, constitute only a I small fraction of a system of interest, they provide geochemical and geological information out of proportion to their abundance. There are several reasons for this. First, variations in the concentrations of many trace elements are much larger than variations in the concentrations of major components, of- ten by many orders of magnitude. Second, in any system there are far more trace elements than major elements. In most geochemical systems, there are 10 or fewer major components that together account for 99% or more of the system. This leaves 80 trace elements. Each element has chemical properties that are to some degree unique, hence there is unique geochemical information contained in the varia- tion of concentration for each element. Thus the 80 trace elements always contain information not available from the variations in the concentrations of major elements. Third, the range in behavior of trace elements is large and collectively they are sensitive to processes to which major elements are in- sensitive. One example is the depth at which partial melting occurs in the mantle. When the mantle melts, it produces melts whose composition is only weakly dependent on pressure, i.e., it always pro- duces basalt. Certain trace elements, however, are highly sensitive to the depth of melting (because the phase assemblages are functions of pressure). -

Significant Zr Isotope Variations in Single Zircon Grains Recording Magma Evolution History
Significant Zr isotope variations in single zircon grains recording magma evolution history Jing-Liang Guoa,b,1, Zaicong Wanga,1, Wen Zhanga, Frédéric Moyniera,c, Dandan Cuia, Zhaochu Hua, and Mihai N. Duceab,d aState Key Laboratory of Geological Processes and Mineral Resources, School of Earth Sciences, China University of Geosciences, Wuhan 430074, China; bDepartment of Geosciences, University of Arizona, Tucson, AZ 85718; cInstitut de Physique du Globe de Paris, Université de Paris, CNRS, 75238 Paris Cedex 05, France; and dFaculty of Geology and Geophysics, University of Bucharest, 010041 Bucharest, Romania Edited by Bruce Watson, Rensselaer Polytechnic Institute, Troy, NY, and approved July 20, 2020 (received for review February 27, 2020) Zircons widely occur in magmatic rocks and often display internal differentiation processes of the silicate Earth (17, 18), the Zr zonation finely recording the magmatic history. Here, we presented isotopes of zircon might also record related messages. in situ high-precision (2SD <0.15‰ for δ94Zr) and high–spatial-resolution Recent developments of bulk and in situ analytical methods (20 μm) stable Zr isotope compositions of magmatic zircons in a have permitted studies on mass-dependent Zr isotope fraction- suite of calc-alkaline plutonic rocks from the juvenile part of the ation in terrestrial samples, including zircons (15, 19–24). They Gangdese arc, southern Tibet. These zircon grains are internally show the great potential of Zr isotope in tracing magmatic zoned with Zr isotopically light cores and increasingly heavier processes. However, available data have been rather limited and rims. Our data suggest the preferential incorporation of lighter resulted in very controversial interpretation (21, 22), such as for Zr isotopes in zircon from the melt, which would drive the residual fractionation factor α (whether greater or smaller than 1). -

Mineral Potential for Incompatible Element Deposits Hosted In
Prepared in cooperation with the Ministry of Petroleum, Energy and Mines, Islamic Republic of Mauritania Second Projet de Renforcement Institutionnel du Secteur Minier de la République Islamique de Mauritanie (PRISM-II) Mineral Potential for Incompatible Element Deposits Hosted in Pegmatites, Alkaline Rocks, and Carbonatites in the Islamic Republic of Mauritania: Phase V, Deliverable 87 By Cliff D. Taylor and Stuart A. Giles Open-File Report 2013–1280 Chapter Q U.S. Department of the Interior U.S. Geological Survey U.S. Department of the Interior SALLY JEWELL, Secretary U.S. Geological Survey Suzette M. Kimball, Acting Director U.S. Geological Survey, Reston, Virginia: 2015 For more information on the USGS—the Federal source for science about the Earth, its natural and living resources, natural hazards, and the environment—visit http://www.usgs.gov or call 1–888–ASK–USGS For an overview of USGS information products, including maps, imagery, and publications, visit http://www.usgs.gov/pubprod To order this and other USGS information products, visit http://store.usgs.gov Suggested citation: Taylor, C.D., and Giles, S.A., 2015, Mineral potential for incompatible element deposits hosted in pegmatites, alkaline rocks, and carbonatites in the Islamic Republic of Mauritania (phase V, deliverable 87), chap. Q of Taylor, C.D., ed., Second projet de renforcement institutionnel du secteur minier de la République Islamique de Mauritanie (PRISM-II): U.S. Geological Survey Open-File Report 2013‒1280-Q, 41 p., http://dx.doi.org/10.3133/ofr20131280. [In English and French.] Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. -

Melting Depths and Mantle Heterogeneity Beneath Hawaii and the East Pacific Rise
JOURNAL OF GEOPHYSICAL RESEARCH, VOL. 104, NO. B2, PAGES 2817–2829, FEBRUARY 10, 1999 Melting depths and mantle heterogeneity beneath Hawaii and the East Pacific Rise: Constraints from Na/Ti and rare earth element ratios Keith Putirka Lawrence Livermore National Laboratory, Livermore, California. Abstract. Mantle melting calculations are presented that place constraints on the mineralogy of the basalt source region and partial melting depths for oceanic basalts. Melting depths are obtained from pressure-sensitive mineral-melt partition coefficients for Na, Ti, Hf, and the rare earth elements (REE). Melting depths are estimated by comparing model aggregate melt compositions to natural basalts from Hawaii and the East Pacific Rise (EPR). Variations in melting depths in a peridotite mantle are sufficient to yield observed differences in Na/Ti, Lu/Hf, and Sm/Yb between Hawaii and the EPR. Initial melting depths of 95–120 km are calculated for EPR basalts, while melting depths of 200–400 km are calculated for Hawaii, indicating a mantle that is 3008C hotter at Hawaii. Some isotope ratios at Hawaii are correlated with Na/Ti, indicating vertical stratification to isotopic heterogeneity in the mantle; similar comparisons involving EPR lavas support a layered mantle model. Abundances of Na, Ti, and REE indicate that garnet pyroxenite and eclogite are unlikely source components at Hawaii and may be unnecessary at the EPR. The result that some geochemical features of oceanic lavas appear to require only minor variations in mantle mineral proportions (2% or less) may have important implications regarding the efficiency of mantle mixing. Heterogeneity required by isotopic studies might be accompanied by only subtle differences in bulk composition, and material that is recycled at subduction zones might not persist as mineralogically distinct mantle components. -

Nevado De Longaví Volcano (Chilean Andes, 36.2 ˚S): Adakitic Magmas Formed by Crystal Fractionation from Hydrous Basalts
Geophysical Research Abstracts, Vol. 7, 01205, 2005 SRef-ID: 1607-7962/gra/EGU05-A-01205 © European Geosciences Union 2005 Nevado de Longaví Volcano (Chilean Andes, 36.2 ˚S): adakitic magmas formed by crystal fractionation from hydrous basalts C. Rodríguez, D. Sellés, M.A. Dungan Université de Genève, Département de Minéralogie. 13 Rue des Maraîchers, 1205 Geneva, Switzerland. The formation of adakites by partial melting of oceanic lithosphere is limited to re- gions where young, hot slabs are subducted. Adakitic melts also occur in continental arcs related to subduction of cold oceanic lithosphere, where they have been explained in terms of remelting of basaltic material underplated at the base of thickened orogenic crust (e.g., Petford et al., 1996; Kay and Mpodozis, 2002) or as the result of modifi- cation of the sub-arc mantle related to tectonic erosion of the forearc crust (e.g. Kay et al., 2004 in press). The only occurrence of Quaternary magmas in the accessible Andean Southern Volcanic Zone (SVZ 33-41˚ S) with an unequivocal adakitic signa- ture is at Nevado de Longaví (NLV; 36˚12’S – 71˚10’ W; Sellés et al. 2004), which is located to the south of the region that has been strongly affected by Tertiary crustal shortening and thickening, and associated eastward arc migration. We propose that the generation of adakitic melts at NLV has occurred via fractional crystallization of water-rich basalts. The unusual chemistry and unusually high modal abundance of hornblende in dacitic magmas at NLV appears to be ultimately related to an excep- tionally high fluid-flux from the subducted Mocha Fracture Zone (Eocene-age Nazca plate), which projects beneath NLV, and which is inferred to have generated water-rich but incompatible element-poor basalts through flux-melting.