Correlation Between Deletion Patterns of SMN and NAIP Genes and the Clinical Features of Spinal Muscular Atrophy in Indian Patients
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
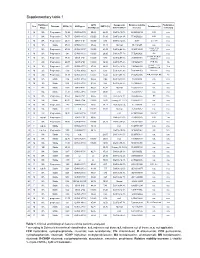
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

ATP-Binding and Hydrolysis in Inflammasome Activation
molecules Review ATP-Binding and Hydrolysis in Inflammasome Activation Christina F. Sandall, Bjoern K. Ziehr and Justin A. MacDonald * Department of Biochemistry & Molecular Biology, Cumming School of Medicine, University of Calgary, 3280 Hospital Drive NW, Calgary, AB T2N 4Z6, Canada; [email protected] (C.F.S.); [email protected] (B.K.Z.) * Correspondence: [email protected]; Tel.: +1-403-210-8433 Academic Editor: Massimo Bertinaria Received: 15 September 2020; Accepted: 3 October 2020; Published: 7 October 2020 Abstract: The prototypical model for NOD-like receptor (NLR) inflammasome assembly includes nucleotide-dependent activation of the NLR downstream of pathogen- or danger-associated molecular pattern (PAMP or DAMP) recognition, followed by nucleation of hetero-oligomeric platforms that lie upstream of inflammatory responses associated with innate immunity. As members of the STAND ATPases, the NLRs are generally thought to share a similar model of ATP-dependent activation and effect. However, recent observations have challenged this paradigm to reveal novel and complex biochemical processes to discern NLRs from other STAND proteins. In this review, we highlight past findings that identify the regulatory importance of conserved ATP-binding and hydrolysis motifs within the nucleotide-binding NACHT domain of NLRs and explore recent breakthroughs that generate connections between NLR protein structure and function. Indeed, newly deposited NLR structures for NLRC4 and NLRP3 have provided unique perspectives on the ATP-dependency of inflammasome activation. Novel molecular dynamic simulations of NLRP3 examined the active site of ADP- and ATP-bound models. The findings support distinctions in nucleotide-binding domain topology with occupancy of ATP or ADP that are in turn disseminated on to the global protein structure. -

NLR Members in Inflammation-Associated
Cellular & Molecular Immunology (2017) 14, 403–405 & 2017 CSI and USTC All rights reserved 2042-0226/17 $32.00 www.nature.com/cmi RESEARCH HIGHTLIGHT NLR members in inflammation-associated carcinogenesis Ha Zhu1,2 and Xuetao Cao1,2,3 Cellular & Molecular Immunology (2017) 14, 403–405; doi:10.1038/cmi.2017.14; published online 3 April 2017 hronic inflammation is regarded as an impor- nucleotide-binding and oligomerization domain IL-2,8 and NAIP was found to regulate the STAT3 Ctant factor in cancer progression. In addition (NOD)-like receptors (NLRs). TLRs and CLRs are pathway independent of inflammasome formation.9 to the immune surveillance function in the early located in the plasma membranes, whereas RLRs, The AOM/DSS model is the most popular model stage of tumorigenesis, inflammation is also known ALRs and NLRs are intracellular PRRs.3 Unlike used to study the function of NLRs in fl fl as one of the hallmarks of cancer and can supply other families that have been shown to bind their in ammation-associated carcinogenesis. In amma- the tumor microenvironment with bioactive mole- specific cognate ligands, the distinct ligands for somes initiated by NLRs or AIM2 have been widely cules and favor the development of other hallmarks NLRs are still unknown. In fact, mounting evidence reported to participate in the maintenance of 10,11 Nlrp3 Nlrp6 of cancer, such as genetic instability and angiogen- suggests that NLRs function as cytoplasmic sensors intestinal homeostasis. -/-, -/-, Nlrc4 Nlrp1 Nlrx1 Nlrp12 esis. Moreover, inflammation contributes to the and participate in modulating TLR, RLR and CLR -/-, -/-, -/- and -/- mice are 4 more susceptible to AOM/DSS-induced colorectal changing tumor microenvironment by altering signaling pathways. -

Apoptosis Ligand-Induced Enhanced Resistance to Fas/Fas
Theileria parva-Transformed T Cells Show Enhanced Resistance to Fas/Fas Ligand-Induced Apoptosis This information is current as Peter Küenzi, Pascal Schneider and Dirk A. E. Dobbelaere of October 2, 2021. J Immunol 2003; 171:1224-1231; ; doi: 10.4049/jimmunol.171.3.1224 http://www.jimmunol.org/content/171/3/1224 Downloaded from References This article cites 69 articles, 29 of which you can access for free at: http://www.jimmunol.org/content/171/3/1224.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on October 2, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2003 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Theileria parva-Transformed T Cells Show Enhanced Resistance to Fas/Fas Ligand-Induced Apoptosis1 Peter Ku¨enzi,2* Pascal Schneider,† and Dirk A. E. Dobbelaere3* Lymphocyte homeostasis is regulated by mechanisms that control lymphocyte proliferation and apoptosis. Activation-induced cell death is mediated by the expression of death ligands and receptors, which, when triggered, activate an apoptotic cascade. -

Techniques for Immune Function Analysis Application Handbook 1St Edition
Techniques for Immune Function Analysis Application Handbook 1st Edition BD Biosciences For additional information please access the Immune Function Homepage at www.bdbiosciences.com/immune_function For Research Use Only. Not for use in diagnostic or therapeutic procedures. Purchase does not include or carry any right to resell or transfer this product either as a stand-alone product or as a component of another product. Any use of this product other than the permitted use without the express written authorization of Becton Dickinson and Company is strictly prohibited. All applications are either tested in-house or reported in the literature. See Technical Data Sheets for details. BD, BD Logo and all other trademarks are the property of Becton, Dickinson and Company. ©2003 BD Table of Contents Preface . 4 Chapter 1: Immunofluorescent Staining of Cell Surface Molecules for Flow Cytometric Analysis . 9 Chapter 2: BD™ Cytometric Bead Array (CBA) Multiplexing Assays . 35 Chapter 3: BD™ DimerX MHC:Ig Proteins for the Analysis of Antigen-specific T Cells. 51 Chapter 4: Immunofluorescent Staining of Intracellular Molecules for Flow Cytometric Analysis . 61 Chapter 5: BD FastImmune™ Cytokine Flow Cytometry. 85 Chapter 6: BD™ ELISPOT Assays for Cells That Secrete Biological Response Modifiers . 109 Chapter 7: ELISA for Specifically Measuring the Levels of Cytokines, Chemokines, Inflammatory Mediators and their Receptors . 125 Chapter 8: BD OptEIA™ ELISA Sets and Kits for Quantitation of Analytes in Serum, Plasma, and Cell Culture Supernatants. 143 Chapter 9: BrdU Staining and Multiparameter Flow Cytometric Analysis of the Cell Cycle . 155 Chapter 10: Cell-based Assays for Biological Response Modifiers . 177 Chapter 11: BD RiboQuant™ Multi-Probe RNase Protection Assay System . -

Molecular Genetics of Spinal Muscular Atrophy: Contribution of the NAIP Gene to Clinical Severity
Kobe J. Med. Sci. 48, 25/31 February 2002 Molecular Genetics of Spinal Muscular Atrophy: Contribution of the NAIP Gene to Clinical Severity TOMOKO AKUTSU1*, HISAHIDE NISHIO2, KIMIAKI SUMINO2, YASUHIRO TAKESHIMA1, SYUICHI TSUNEISHI1, HIROKO WADA1, SATOSHI TAKADA3, MASAFUMI MATSUO1, 4, and HAJIME NAKAMURA1 Division of Pediatrics, Department of Development and Aging1, Division of Public Health, Department of Environmental Health and Safety2, Kobe University Graduate School of Medicine Faculty of Health Science, Kobe University School of Medicine, Kobe 654-0142, Japan3 Division of Molecular Medicine, Department of International and Environmental Medical Sciences, Kobe University Graduate School of Medicine4 Received 5 February 2002/ Accepted 19 February 2002 Key words: spinal muscular atrophy; the SMN1 gene; the NAIP gene; the p44t gene Spinal muscular atrophy (SMA) is one of the most common autosomal recessive disorders characterized by degeneration of anterior horn cells in the spinal cord, and leads to progressive muscular weakness and atrophy. At least three SMA-related genes have been identified: SMN1, NAIP and p44t. We analyzed these genes in 32 SMA patients and found that the SMN1 gene was deleted in 30 of 32 patients (94 %), irrespective of clinical type. The NAIP gene was deleted in 6 patients and its deletion rate was higher in type I patients than that in typeⅡ or Ⅲ. Further, in type I patients lacking the NAIP gene, deterioration in their respiratory function is more rapid than in those type I patients retaining the NAIP gene. Since complete p44t deletion was observed in only 3 patients, the correlation between the p44t deletion and severity of SMA remained ambiguous. -

The Role of Nucleolin in B-Cell Lymphomas and Fas-Mediated Apoptotic Signaling
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 5-2013 The Role of Nucleolin in B-cell Lymphomas and Fas-Mediated Apoptotic Signaling Jillian F. Wise Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Cancer Biology Commons, and the Medicine and Health Sciences Commons Recommended Citation Wise, Jillian F., "The Role of Nucleolin in B-cell Lymphomas and Fas-Mediated Apoptotic Signaling" (2013). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 339. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/339 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. The Role of Nucleolin in B-cell Lymphomas and Fas-Mediated Apoptotic Signaling by Jillian F Wise, BS Approved: ___________________________________________________ Felipe -

Bacterial Detection by NAIP/NLRC4 Elicits Prompt Contractions of Intestinal Epithelial Cell Layers
Bacterial detection by NAIP/NLRC4 elicits prompt contractions of intestinal epithelial cell layers Pilar Samperio Ventayola, Petra Geisera, Maria Letizia Di Martinoa, Alexandra Florbranta, Stefan A. Fattingera,b, Naemi Walderb, Eduardo Simac, Feng Shaod, Nelson O. Gekarae, Magnus Sundbomc, Wolf-Dietrich Hardtb, Dominic-Luc Webbf, Per M. Hellströmf, Jens Erikssona, and Mikael E. Sellina,1 aScience for Life Laboratory, Department of Medical Biochemistry and Microbiology, Uppsala University, 75123 Uppsala, Sweden; bInstitute of Microbiology, Department of Biology, ETH Zürich, 8093 Zürich, Switzerland; cDepartment of Surgical Sciences, Uppsala University, 75185 Uppsala, Sweden; dNational Institute of Biological Sciences, 102206 Beijing, China; eDepartment of Molecular Biosciences, The Wenner-Gren Institute, Stockholm University, Stockholm 10691, Sweden; and fDepartment of Medical Sciences, Gastroenterology and Hepatology Unit, Uppsala University, 75185 Uppsala, Sweden Edited by Ralph R. Isberg, Tufts University School of Medicine, Boston, MA, and approved February 24, 2021 (received for review July 3, 2020) The gut epithelium serves to maximize the surface for nutrient and form inflammasomes in the cytosol] (7–9). Epithelial recognition fluid uptake, but at the same time must provide a tight barrier to of pathogens through PRRs elicits a panel of countermeasures, pathogens and remove damaged intestinal epithelial cells (IECs) including production of proinflammatory cytokines, chemokines, without jeopardizing barrier integrity. How the epithelium coor- lipids (10–13), secretion of antimicrobial peptides (11, 14), and dinates these tasks remains a question of significant interest. We the death and expulsion of infected IECs into the lumen (12, 15, used imaging and an optical flow analysis pipeline to study the 16). An intricate cross-talk exists between IECs and immune cells dynamicity of untransformed murine and human intestinal epi- residing in the underlying lamina propria (17). -

Genotype-Phenotype Correlation of SMN Locus Genes in Spinal Muscular Atrophy Patients from India
EXPERIMENTAL and MOLECULAR MEDICINE, Vol. 37, No. 3, 147-154, June 2005 Genotype-Phenotype correlation of SMN locus genes in spinal muscular atrophy patients from India Akanchha Kesari1, M Mohammed Idris2, Introduction Giri Raj Chandak2 and Balraj Mittal1,3 Proximal spinal muscular atrophy (SMA) is an auto- 1 somal recessive neuromuscular disorder, characteri- Department of Genetics zed by destruction of α-motor neurons of anterior horn Sanjay Gandhi Postgraduate cells, which leads to symmetrical muscle weakness Institute of Medical Sciences and atrophy. It has an estimated incidence of 1/ Raebareli Road, Lucknow-226 014-India 10,000 live births (Panigrahi et al., 2002), and a 2Centre for Cellular and Molecular Biology, Uppal Road, carrier frequency of 1/50. Clinically proximal SMA is Hyderabad-500 007-India further divided into four types based on the age of 3Corresponding author: Tel, 91-522-2668005; onset and severity of the clinical course. Childhood Fax, 91-522-2668973; E-mail, [email protected] onset SMAs can be classified into three types (Type I-III) (Biros and Forrest, 1999). Type I is the most severe form having symptoms of SMA before 6 Accepted 25 March 2005 months. Type II is an intermediate form with onset before the age of 18 months. Type III is a relatively Abbreviations: BTFp44, basal transcription factor subunit p44; milder, chronic form with onset after the age of 18 NAIP, neuronal apoptosis inhibitory protein; SMA, spinal muscular months. Type IV SMA is an adult form with onset atrophy; SMN, survival of motor neuron gene after 30 years of age with variable severity but normal life span. -

The Role of Apoptosis in the Pathology of Pancreatic Cancer
Cancers 2011, 3, 1-16; doi:10.3390/cancers3010001 OPEN ACCESS cancers ISSN 2072-6694 www.mdpi.com/journal/cancers Review The Role of Apoptosis in the Pathology of Pancreatic Cancer Nicole Samm, Kristin Werner, Felix Rückert, Hans Detlev Saeger, Robert Grützmann and Christian Pilarsky * Department of Visceral-, Thoracic- and Vascular-Surgery, University Hospital Dresden, Dresden, Germany; E-Mails: [email protected] (N.S.); [email protected] (K.W.); [email protected] (F.R.); [email protected] (H.D.S.); [email protected] (R.G.) * Author to whom correspondence should be addressed; Tel.: +49 351 458 166 07; Fax: +49 351 449 210 303; E-Mail: [email protected]. Received: 11 November 2010; in revised form: 14 December 2010 / Accepted: 21 December 2010 / Published: 23 December 2010 Abstract: Pancreatic cancer is a disease with high resistance to most common therapies and therefore has a poor prognosis, which is partly due to a lack of reaction to apoptotic stimuli. Signal transduction of such stimuli includes a death receptor-mediated extrinsic pathway as well as an intrinsic pathway linked to the mitochondria. Defects in apoptotic pathways and the deregulation of apoptotic proteins, such as Survivin, Bcl-2, Bcl-xL and Mcl-1, play decisive roles in the development of pancreatic cancer. Investigation of the molecular mechanism allowing tumors to resist apoptotic cell death would lead to an improved understanding of the physiology and the development of new molecular strategies in pancreatic cancer. Keywords: apoptosis; signal transduction pathway; pancreatic cancer 1. Introduction Pancreatic cancer is one of the most malignant and aggressive types of cancer in humans and carries a very poor prognosis. -

Salmonella Flagellin Activates NAIP/NLRC4 and Canonical NLRP3 Inflammasomes in Human Macrophages
Salmonella Flagellin Activates NAIP/NLRC4 and Canonical NLRP3 Inflammasomes in Human Macrophages This information is current as Anna M. Gram, John A. Wright, Robert J. Pickering, of September 28, 2021. Nathaniel L. Lam, Lee M. Booty, Steve J. Webster and Clare E. Bryant J Immunol published online 30 December 2020 http://www.jimmunol.org/content/early/2020/12/29/jimmun ol.2000382 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2020/12/29/jimmunol.200038 Material 2.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 28, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Author Choice Freely available online through The Journal of Immunology Author Choice option Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2020 The Authors All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published December 30, 2020, doi:10.4049/jimmunol.2000382 The Journal of Immunology Salmonella Flagellin Activates NAIP/NLRC4 and Canonical NLRP3 Inflammasomes in Human Macrophages Anna M. Gram,* John A. Wright,*,1 Robert J. Pickering,* Nathaniel L. Lam,*,† Lee M. -

The Hippocampal Neurons of Neuronal Apoptosis Inhibitory Protein 1 (NAIP1)-Deleted Mice Display Increased Vulnerability to Kainic Acid-Induced Injury
The hippocampal neurons of neuronal apoptosis inhibitory protein 1 (NAIP1)-deleted mice display increased vulnerability to kainic acid-induced injury Martin Holcik*†, Charlie S. Thompson†, Zahra Yaraghi*, Charles A. Lefebvre†, Alex E. MacKenzie*†‡, and Robert G. Korneluk*†§ *Solange Gauthier Karsh Molecular Genetics Laboratory, Children’s Hospital of Eastern Ontario Research Institute, Ottawa, ON Canada K1H 8L1; and §Department of Biochemistry, Microbiology, and Immunology, and †Apoptogen Incorporated, University of Ottawa, Ottawa, ON Canada K1H 8M5 Edited by W. Maxwell Cowan, Howard Hughes Medical Institute, Chevy Chase, MD, and approved December 17, 1999 (received for review October 29, 1999) The neuronal apoptosis inhibitory protein (NAIP) is a member of a transient forebrain ischemia, suggesting that NAIP plays a key novel family of inhibitor of apoptosis (IAP) proteins. The IAP genes role in neuroprotection (13). are highly conserved from baculovirus to metazoans and suppress The role of the IAP genes in the control and modulation of apoptosis induced by a variety of triggers both in vitro and in vivo. programmed cell death has been mainly inferred from the in vitro Here we describe the generation and characterization of mice with experimentation. The in vivo physiological roles of the endoge- the targeted deletion of NAIP1. We demonstrate that the NAIP1- nous IAP genes are less clear due to the lack of data from the deleted mice develop normally. However, the survival of pyramidal knock-out models. In the present paper, we describe the gener- neurons in the hippocampus after kainic acid-induced limbic sei- ation and characterization of the NAIP1 knock-out mice. We zures is greatly reduced in the NAIP1 knock-out animals.