The Role of Nucleolin in B-Cell Lymphomas and Fas-Mediated Apoptotic Signaling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Folic Acid Antagonists: Antimicrobial and Immunomodulating Mechanisms and Applications
International Journal of Molecular Sciences Review Folic Acid Antagonists: Antimicrobial and Immunomodulating Mechanisms and Applications Daniel Fernández-Villa 1, Maria Rosa Aguilar 1,2 and Luis Rojo 1,2,* 1 Instituto de Ciencia y Tecnología de Polímeros, Consejo Superior de Investigaciones Científicas, CSIC, 28006 Madrid, Spain; [email protected] (D.F.-V.); [email protected] (M.R.A.) 2 Consorcio Centro de Investigación Biomédica en Red de Bioingeniería, Biomateriales y Nanomedicina, 28029 Madrid, Spain * Correspondence: [email protected]; Tel.: +34-915-622-900 Received: 18 September 2019; Accepted: 7 October 2019; Published: 9 October 2019 Abstract: Bacterial, protozoan and other microbial infections share an accelerated metabolic rate. In order to ensure a proper functioning of cell replication and proteins and nucleic acids synthesis processes, folate metabolism rate is also increased in these cases. For this reason, folic acid antagonists have been used since their discovery to treat different kinds of microbial infections, taking advantage of this metabolic difference when compared with human cells. However, resistances to these compounds have emerged since then and only combined therapies are currently used in clinic. In addition, some of these compounds have been found to have an immunomodulatory behavior that allows clinicians using them as anti-inflammatory or immunosuppressive drugs. Therefore, the aim of this review is to provide an updated state-of-the-art on the use of antifolates as antibacterial and immunomodulating agents in the clinical setting, as well as to present their action mechanisms and currently investigated biomedical applications. Keywords: folic acid antagonists; antifolates; antibiotics; antibacterials; immunomodulation; sulfonamides; antimalarial 1. -

A National Strategy for the Elimination of Hepatitis B and C: Phase Two Report
THE NATIONAL ACADEMIES PRESS This PDF is available at http://www.nap.edu/24731 SHARE A National Strategy for the Elimination of Hepatitis B and C: Phase Two Report DETAILS 296 pages | 6 x 9 | PAPERBACK ISBN 978-0-309-45729-3 | DOI: 10.17226/24731 CONTRIBUTORS GET THIS BOOK Gillian J. Buckley and Brian L. Strom, Editors; Committee on a National Strategy for the Elimination of Hepatitis B and C; Board on Population Health and Public Health Practice; Health and Medicine FIND RELATED TITLES Division; National Academies of Sciences, Engineering, and Medicine Visit the National Academies Press at NAP.edu and login or register to get: – Access to free PDF downloads of thousands of scientific reports – 10% off the price of print titles – Email or social media notifications of new titles related to your interests – Special offers and discounts Distribution, posting, or copying of this PDF is strictly prohibited without written permission of the National Academies Press. (Request Permission) Unless otherwise indicated, all materials in this PDF are copyrighted by the National Academy of Sciences. Copyright © National Academy of Sciences. All rights reserved. A National Strategy for the Elimination of Hepatitis B and C: Phase Two Report Gillian J. Buckley and Brian L. Strom, Editors Committee on a National Strategy for the Elimination of Hepatitis B and C Board on Population Health and Public Health Practice Health and Medicine Division A Report of Copyright © National Academy of Sciences. All rights reserved. A National Strategy for the Elimination of Hepatitis B and C: Phase Two Report THE NATIONAL ACADEMIES PRESS 500 Fifth Street, NW Washington, DC 20001 This activity was supported by the American Association for the Study of Liver Diseases, the Infectious Diseases Society of America, the National Viral Hepatitis Roundtable, and the U.S. -

IHC) Outreach Services
IIImmunohistochemistry (IHC) Outreach Services Note type of fixative used if not neutral buffered formalin. Note type of tissue/specimen Unless specified otherwise, positive and negative controls react satisfactorily. Available Chromogen – All markers have been validated with 3,3’-Diaminobenzidine Tetrahydrochloride (DAB) which results in a brown/black precipitate. DAB is the routine chromogen. In addition, some markers have also been validated using the Fast Red (RED), which results in a red precipitate. If available with both chromogens and one is not selected, the def ault will be the DAB chromogen. Antibody Common Applications St aining Charac teristi cs Actin (muscl e sp eci fic ) Smoo th, ske letal & ca rdiac musc le Cytoplasm ic Actin (smoo th muscle ) Smoo th muscl e an d myoep itheli al cel ls Cytoplasm ic and memb rane ALK Pr otein ALK1 posi tive lymp homa s Cytoplasm ic and/or nuclea r Alph a-1-Antitr ypsi n Demo nstr ates A-1-AT in liver Cytoplasm ic (A-1-AT) Bcl-2 Oncop rotein Foll icula r lymph oma an d so ft tiss ue Cytoplasm ic tumors Bcl-6 Foll icula r lymph oma Nucl ear Ber-EP 4, Epithelia l Antige n Aden oca rcin oma vs. meso the liom a Memb rane and cytoplasm ic. The and epithelial tumors membrane staining is preferentially basolateral. Beta-Amyloid Pos t mo rt em dia gnos is of demen tia E xtr acel lular de pos ition (am yloid plaques), vascular deposition (amyloid angiopathy) BRS T-3, (B72.3) Aden oca rcin oma vs. -

Pharmacology
Step I Pharmacology 'WSMU~ is a ~aint program 0~the lfederatlan° a"• State Meaieal Boaras a~the I!Jntitea States • lne, and the Natianal Baara a1 Medieal Examiners. USMLE·Step 1 Pharmacology Lecture Notes 2006-2007 Edition KAPLA~. I meulca • USMLE is a joint program of the Federation of State Medical Boards of the United States, Inc. and the National Board of Medical Examiners. ©2006 Kaplan, Inc. All rights reserved. No part of this book may be reproduced in any form, by photostat, microfilm, xerography or any other means, or incorporated into any information retrieval system, electronic or mechanical, without the written permission of Kaplan, Inc. Not for resale. Author Lionel P. Rayman, Pharm.D., Ph.D. Department of Pathology Forensic Toxicology Laboratory University of Miami School of Medicine Miami, FL Contributing Authors Director of Medical Curriculum Sonia Reichert, M.D. Craig Davis, Ph.D. Associate Professor Editorial Director University of South Carolina School of Medicine Department of Pharmacology, Physiology, and Neuroscience Kathlyn McGreevy Columbia, SC Production Manager Maris Victor Nora, Pharm.D., Ph.D. Michael Wolff Associate Professor Rush Medical College Production Editor Chicago.Tl. William Ng Anthony Trevor, Ph.D. Cover Design Professor Emeritus Joanna Myllo Department of Cellular and Molecular Pharmacology University of California Cover Art San Francisco, CA Christine Schaar Steven R. Harris, Ph.D. Associate Dean for Basic Sciences Associate Professor of Pharmacology Pikeville College School of Osteopathic Medicine -

Key Enzymes in Cancer: Mechanism of Action and Inhibition with Anticancer Agents
University of Texas Rio Grande Valley ScholarWorks @ UTRGV Chemistry Faculty Publications and Presentations College of Sciences 2018 Key Enzymes in Cancer: Mechanism of Action and Inhibition With Anticancer Agents Debasish Bandyopadhyay The University of Texas Rio Grande Valley, [email protected] Gabriel Lopez The University of Texas Rio Grande Valley Stephanie Cantu The University of Texas Rio Grande Valley Samantha Balboa The University of Texas Rio Grande Valley Annabel Garcia The University of Texas Rio Grande Valley See next page for additional authors Follow this and additional works at: https://scholarworks.utrgv.edu/chem_fac Part of the Chemistry Commons, and the Life Sciences Commons Recommended Citation Debasish Bandyopadhyay, Gabriel Lopez, Stephanie Cantu, Samantha Balboa, Annabel Garcia, Christina Silva, Diandra Valdes. In Chemistry Research and Applications (Vol. 2): Organic and Medicinal Chemistry, Chapter 9; Key Enzymes in Cancer: Mechanism of Action and Inhibition with Anticancer Agents. 2018, Nova Science Publishers, Inc., Hauppauge, New York, USA (ISBN: 978-1-53614-855-8). This Book is brought to you for free and open access by the College of Sciences at ScholarWorks @ UTRGV. It has been accepted for inclusion in Chemistry Faculty Publications and Presentations by an authorized administrator of ScholarWorks @ UTRGV. For more information, please contact [email protected], [email protected]. Authors Debasish Bandyopadhyay, Gabriel Lopez, Stephanie Cantu, Samantha Balboa, Annabel Garcia, Christina Silva, and Diandra Valdes This book is available at ScholarWorks @ UTRGV: https://scholarworks.utrgv.edu/chem_fac/131 In: Organic and Medicinal Chemistry, Volume 2 ISBN: 978-1-53614-855-8 Editor: Bimal Krishna Banik © 2019 Nova Science Publishers, Inc. -
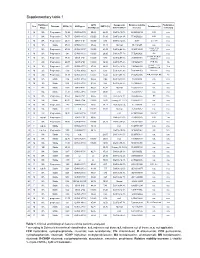
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

Nucleolin and Its Role in Ribosomal Biogenesis
NUCLEOLIN: A NUCLEOLAR RNA-BINDING PROTEIN INVOLVED IN RIBOSOME BIOGENESIS Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf vorgelegt von Julia Fremerey aus Hamburg Düsseldorf, April 2016 2 Gedruckt mit der Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf Referent: Prof. Dr. A. Borkhardt Korreferent: Prof. Dr. H. Schwender Tag der mündlichen Prüfung: 20.07.2016 3 Die vorgelegte Arbeit wurde von Juli 2012 bis März 2016 in der Klinik für Kinder- Onkologie, -Hämatologie und Klinische Immunologie des Universitätsklinikums Düsseldorf unter Anleitung von Prof. Dr. A. Borkhardt und in Kooperation mit dem ‚Laboratory of RNA Molecular Biology‘ an der Rockefeller Universität unter Anleitung von Prof. Dr. T. Tuschl angefertigt. 4 Dedicated to my family TABLE OF CONTENTS 5 TABLE OF CONTENTS TABLE OF CONTENTS ............................................................................................... 5 LIST OF FIGURES ......................................................................................................10 LIST OF TABLES .......................................................................................................12 ABBREVIATION .........................................................................................................13 ABSTRACT ................................................................................................................19 ZUSAMMENFASSUNG -

ATP-Binding and Hydrolysis in Inflammasome Activation
molecules Review ATP-Binding and Hydrolysis in Inflammasome Activation Christina F. Sandall, Bjoern K. Ziehr and Justin A. MacDonald * Department of Biochemistry & Molecular Biology, Cumming School of Medicine, University of Calgary, 3280 Hospital Drive NW, Calgary, AB T2N 4Z6, Canada; [email protected] (C.F.S.); [email protected] (B.K.Z.) * Correspondence: [email protected]; Tel.: +1-403-210-8433 Academic Editor: Massimo Bertinaria Received: 15 September 2020; Accepted: 3 October 2020; Published: 7 October 2020 Abstract: The prototypical model for NOD-like receptor (NLR) inflammasome assembly includes nucleotide-dependent activation of the NLR downstream of pathogen- or danger-associated molecular pattern (PAMP or DAMP) recognition, followed by nucleation of hetero-oligomeric platforms that lie upstream of inflammatory responses associated with innate immunity. As members of the STAND ATPases, the NLRs are generally thought to share a similar model of ATP-dependent activation and effect. However, recent observations have challenged this paradigm to reveal novel and complex biochemical processes to discern NLRs from other STAND proteins. In this review, we highlight past findings that identify the regulatory importance of conserved ATP-binding and hydrolysis motifs within the nucleotide-binding NACHT domain of NLRs and explore recent breakthroughs that generate connections between NLR protein structure and function. Indeed, newly deposited NLR structures for NLRC4 and NLRP3 have provided unique perspectives on the ATP-dependency of inflammasome activation. Novel molecular dynamic simulations of NLRP3 examined the active site of ADP- and ATP-bound models. The findings support distinctions in nucleotide-binding domain topology with occupancy of ATP or ADP that are in turn disseminated on to the global protein structure. -

Cancer Stem Cells and Nucleolin As Drivers of Carcinogenesis
pharmaceuticals Review Cancer Stem Cells and Nucleolin as Drivers of Carcinogenesis Laura Sofia Carvalho 1,Nélio Gonçalves 1 , Nuno André Fonseca 1,2 and João Nuno Moreira 1,3,* 1 CNC—Center for Neurosciences and Cell Biology, Center for Innovative Biomedicine and Biotechnology (CIBB), Faculty of Medicine (Polo 1), University of Coimbra, Rua Larga, 3004-504 Coimbra, Portugal; laurasofi[email protected] (L.S.C.); [email protected] (N.G.); [email protected] (N.A.F.) 2 TREAT U, SA—Parque Industrial de Taveiro, Lote 44, 3045-508 Coimbra, Portugal 3 UC—University of Coimbra, CIBB, Faculty of Pharmacy (FFUC), Pólo das Ciências da Saúde, Azinhaga de Santa Comba, 3000-548 Coimbra, Portugal * Correspondence: [email protected]; Tel.: +351-239-820-190 Abstract: Cancer, one of the most mortal diseases worldwide, is characterized by the gain of specific features and cellular heterogeneity. Clonal evolution is an established theory to explain heterogeneity, but the discovery of cancer stem cells expanded the concept to include the hierarchical growth and plasticity of cancer cells. The activation of epithelial-to-mesenchymal transition and its molecular players are widely correlated with the presence of cancer stem cells in tumors. Moreover, the acquisition of certain oncological features may be partially attributed to alterations in the levels, location or function of nucleolin, a multifunctional protein involved in several cellular processes. This review aims at integrating the established hallmarks of cancer with the plasticity of cancer cells as an emerging hallmark; responsible for tumor heterogeneity; therapy resistance and relapse. The discussion will contextualize the involvement of nucleolin in the establishment of cancer hallmarks and its application as a marker protein for targeted anticancer therapies Keywords: tumor heterogeneity; drug resistance; cancer stem cells; nucleolin; targeted therapies; epithelial-to-mesenchymal transition Citation: Carvalho, L.S.; Gonçalves, N.; Fonseca, N.A.; Moreira, J.N. -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr. -

NLR Members in Inflammation-Associated
Cellular & Molecular Immunology (2017) 14, 403–405 & 2017 CSI and USTC All rights reserved 2042-0226/17 $32.00 www.nature.com/cmi RESEARCH HIGHTLIGHT NLR members in inflammation-associated carcinogenesis Ha Zhu1,2 and Xuetao Cao1,2,3 Cellular & Molecular Immunology (2017) 14, 403–405; doi:10.1038/cmi.2017.14; published online 3 April 2017 hronic inflammation is regarded as an impor- nucleotide-binding and oligomerization domain IL-2,8 and NAIP was found to regulate the STAT3 Ctant factor in cancer progression. In addition (NOD)-like receptors (NLRs). TLRs and CLRs are pathway independent of inflammasome formation.9 to the immune surveillance function in the early located in the plasma membranes, whereas RLRs, The AOM/DSS model is the most popular model stage of tumorigenesis, inflammation is also known ALRs and NLRs are intracellular PRRs.3 Unlike used to study the function of NLRs in fl fl as one of the hallmarks of cancer and can supply other families that have been shown to bind their in ammation-associated carcinogenesis. In amma- the tumor microenvironment with bioactive mole- specific cognate ligands, the distinct ligands for somes initiated by NLRs or AIM2 have been widely cules and favor the development of other hallmarks NLRs are still unknown. In fact, mounting evidence reported to participate in the maintenance of 10,11 Nlrp3 Nlrp6 of cancer, such as genetic instability and angiogen- suggests that NLRs function as cytoplasmic sensors intestinal homeostasis. -/-, -/-, Nlrc4 Nlrp1 Nlrx1 Nlrp12 esis. Moreover, inflammation contributes to the and participate in modulating TLR, RLR and CLR -/-, -/-, -/- and -/- mice are 4 more susceptible to AOM/DSS-induced colorectal changing tumor microenvironment by altering signaling pathways. -

Induces Antigen Presentation in B Cells Cell-Activating Factor of The
B Cell Maturation Antigen, the Receptor for a Proliferation-Inducing Ligand and B Cell-Activating Factor of the TNF Family, Induces Antigen Presentation in B Cells This information is current as of September 27, 2021. Min Yang, Hidenori Hase, Diana Legarda-Addison, Leena Varughese, Brian Seed and Adrian T. Ting J Immunol 2005; 175:2814-2824; ; doi: 10.4049/jimmunol.175.5.2814 http://www.jimmunol.org/content/175/5/2814 Downloaded from References This article cites 54 articles, 36 of which you can access for free at: http://www.jimmunol.org/content/175/5/2814.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 27, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2005 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology B Cell Maturation Antigen, the Receptor for a Proliferation-Inducing Ligand and B Cell-Activating Factor of the TNF Family, Induces Antigen Presentation in B Cells1 Min Yang,* Hidenori Hase,* Diana Legarda-Addison,* Leena Varughese,* Brian Seed,† and Adrian T.