The NOD: a Signaling Module That Regulates Apoptosis and Host Defense Against Pathogens
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Expression of NOD2, NLRP3 and NLRC5 and Renal Injury in Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis
Wang et al. J Transl Med (2019) 17:197 https://doi.org/10.1186/s12967-019-1949-5 Journal of Translational Medicine RESEARCH Open Access The expression of NOD2, NLRP3 and NLRC5 and renal injury in anti-neutrophil cytoplasmic antibody-associated vasculitis Luo‑Yi Wang1,2,3, Xiao‑Jing Sun1,2,3, Min Chen1,2,3* and Ming‑Hui Zhao1,2,3,4 Abstract Background: Nucleotide‑binding oligomerization domain (NOD)‑like receptors (NLRs) are intracellular sensors of pathogens and molecules from damaged cells to regulate the infammatory response in the innate immune system. Emerging evidences suggested a potential role of NLRs in anti‑neutrophil cytoplasmic antibody (ANCA)‑associated vasculitis (AAV). This study aimed to investigate the expression of nucleotide‑binding oligomerization domain con‑ taining protein 2 (NOD2), NOD‑like receptor family pyrin domain containing 3 (NLRP3) and NOD‑like receptor family CARD domain containing 5 (NLRC5) in kidneys of AAV patients, and further explored their associations with clinical and pathological parameters. Methods: Thirty‑four AAV patients in active stage were recruited. Their renal specimens were processed with immu‑ nohistochemistry to assess the expression of three NLRs, and with double immunofuorescence to detect NLRs on intrinsic and infltrating cells. Analysis of gene expression was also adopted in cultured human podocytes. The associa‑ tions between expression of NLRs and clinicopathological parameters were analyzed. Results: The expression of NOD2, NLRP3 and NLRC5 was signifcantly higher in kidneys from AAV patients than those from normal controls, minimal change disease or class IV lupus nephritis. These NLRs co‑localized with podocytes and infltrating infammatory cells. -

Golgi Phosphoprotein 3 Promotes the Proliferation of Gallbladder Carcinoma Cells Via Regulation of the NLRP3 Inflammasome
ONCOLOGY REPORTS 45: 113, 2021 Golgi phosphoprotein 3 promotes the proliferation of gallbladder carcinoma cells via regulation of the NLRP3 inflammasome ZHENCHENG ZHU1,2*, QINGZHOU ZHU1,2*, DONGPING CAI3, LIANG CHEN4, WEIXUAN XIE2, YANG BAI2 and KUNLUN LUO1,2 1Anhui Medical University, Hefei, Anhui 230032; Departments of 2Hepatobiliary Surgery, 3Laboratory and 4Cardiology, The 904th Hospital of Joint Logistic Support Force of PLA, Wuxi, Jiangsu 214044, P.R. China Received January 18, 2021; Accepted April 2, 2021 DOI: 10.3892/or.2021.8064 Abstract. Golgi phosphoprotein 3 (GOLPH3) has been Introduction demonstrated to promote tumor progression in various gastro‑ intestinal malignancies. However, its effects in gallbladder Gallbladder carcinoma (GBC) is a highly malignant tumor carcinoma (GBC) remain unknown. In the present study, the of the biliary system with a median survival time of only expression levels of GOLPH3 and nucleotide‑binding domain 6 months (1‑3). The primary pathological type of GBC leucine‑rich repeat and pyrin domain containing receptor 3 observed in patients is adenocarcinoma. The effects of current (NLRP3) in human GBC tissues were detected by immuno‑ chemotherapeutic regimens are not sufficient for GBC due histochemistry, and the clinical data and survival of these to the lack of effective drugs, making it particularly difficult patients were analyzed. Next, whether GOLPH3 could affect to control the mortality rate of GBC (3,4). Due to the close tumor proliferation via regulation of the NLRP3 inflamma‑ relationship between inflammation and GBC, investigation of some was investigated in vitro. The results demonstrated that inflammatory‑related molecular mechanisms may highlight GOLPH3 could promote GBC cell proliferation, and that it novel specific targets for the treatment of GBC (4). -

Genetic Variation in Pattern-Recognition Receptors and Association with Leprosy 145
DOI: 10.5772/intechopen.73871 ProvisionalChapter chapter 8 Genetic Variation inin Pattern-RecognitionPattern-Recognition ReceptorsReceptors and and Association withwith LeprosyLeprosy Karina Talita de Oliveira Santana JorgeJorge andand Frederico Marianetti SorianiSoriani Additional information isis available atat thethe endend ofof thethe chapterchapter http://dx.doi.org/10.5772/intechopen.73871 Abstract Mycobacterium leprae is a highly infectious and low pathogenic microorganism that is the causal agent of leprosy. The differences in vulnerability to leprosy, the spectral immune response, and the clinical manifestations of this disease are related to different genetic backgrounds among individuals. In this sense, genetic variants, especially in genes related to mycobacteria recognition and host immune response, may be key factors to explain individual susceptibility and resistance to leprosy and their conditions. In this chapter, studies regarding association of genetic variants in pattern-recognition receptors (PRRs) and leprosy will be reviewed revealing the importance of molecules such as Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-containing protein 2 (NOD2) in leprosy initiation and maintenance. Keywords: polymorphisms, pattern-recognition receptors, mycobacterium leprae, leprosy 1. Introduction Leprosy is caused by Mycobacterium leprae, which is an intracellular bacterium with high infectivity and low pathogenicity. It means that there are a large number of people exposed to this pathogen; however, the majority of them are naturally resistant. On the other hand, some people develop leprosy once challenged with M. leprae. The people who develop disease may present different clinical forms of leprosy. Some of them develop a localized disease, named tuberculoid leprosy, with a strong host response, which does not avoid development of nerve injury and physical disabilities. -

Cytotoxic T Cells Class I- Dependent Lymphocyte Killing by NLRC5 Deficiency Selectively Impairs
The Journal of Immunology NLRC5 Deficiency Selectively Impairs MHC Class I- Dependent Lymphocyte Killing by Cytotoxic T Cells Francesco Staehli,* Kristina Ludigs,* Leonhard X. Heinz,† Queralt Seguı´n-Este´vez,‡ Isabel Ferrero,x Marion Braun,x Kate Schroder,{ Manuele Rebsamen,† Aubry Tardivel,* Chantal Mattmann,* H. Robson MacDonald,x Pedro Romero,x Walter Reith,‡ Greta Guarda,*,1 and Ju¨rg Tschopp*,1,2 Nucleotide-binding oligomerization domain-like receptors (NLRs) are intracellular proteins involved in innate-driven inflamma- tory responses. The function of the family member NLR caspase recruitment domain containing protein 5 (NLRC5) remains a matter of debate, particularly with respect to NF-kB activation, type I IFN, and MHC I expression. To address the role of NLRC5, we generated Nlrc5-deficient mice (Nlrc5D/D). In this article we show that these animals exhibit slightly decreased CD8+ T cell percentages, a phenotype compatible with deregulated MHC I expression. Of interest, NLRC5 ablation only mildly affected MHC I expression on APCs and, accordingly, Nlrc5D/D macrophages efficiently primed CD8+ T cells. In contrast, NLRC5 deficiency dramatically impaired basal expression of MHC I in T, NKT, and NK lymphocytes. NLRC5 was sufficient to induce MHC I expression in a human lymphoid cell line, requiring both caspase recruitment and LRR domains. Moreover, endogenous NLRC5 localized to the nucleus and occupied the proximal promoter region of H-2 genes. Consistent with downregulated MHC I expression, the elimination of Nlrc5D/D lymphocytes by cytotoxic T cells was markedly reduced and, in addition, we observed low NLRC5 expression in several murine and human lymphoid-derived tumor cell lines. -

Post-Transcriptional Inhibition of Luciferase Reporter Assays
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 287, NO. 34, pp. 28705–28716, August 17, 2012 © 2012 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A. Post-transcriptional Inhibition of Luciferase Reporter Assays by the Nod-like Receptor Proteins NLRX1 and NLRC3* Received for publication, December 12, 2011, and in revised form, June 18, 2012 Published, JBC Papers in Press, June 20, 2012, DOI 10.1074/jbc.M111.333146 Arthur Ling‡1,2, Fraser Soares‡1,2, David O. Croitoru‡1,3, Ivan Tattoli‡§, Leticia A. M. Carneiro‡4, Michele Boniotto¶, Szilvia Benko‡5, Dana J. Philpott§, and Stephen E. Girardin‡6 From the Departments of ‡Laboratory Medicine and Pathobiology and §Immunology, University of Toronto, Toronto M6G 2T6, Canada, and the ¶Modulation of Innate Immune Response, INSERM U1012, Paris South University School of Medicine, 63, rue Gabriel Peri, 94276 Le Kremlin-Bicêtre, France Background: A number of Nod-like receptors (NLRs) have been shown to inhibit signal transduction pathways using luciferase reporter assays (LRAs). Results: Overexpression of NLRX1 and NLRC3 results in nonspecific post-transcriptional inhibition of LRAs. Conclusion: LRAs are not a reliable technique to assess the inhibitory function of NLRs. Downloaded from Significance: The inhibitory role of NLRs on specific signal transduction pathways needs to be reevaluated. Luciferase reporter assays (LRAs) are widely used to assess the Nod-like receptors (NLRs)7 represent an important class of activity of specific signal transduction pathways. Although pow- intracellular pattern recognition molecules (PRMs), which are erful, rapid and convenient, this technique can also generate implicated in the detection and response to microbe- and dan- www.jbc.org artifactual results, as revealed for instance in the case of high ger-associated molecular patterns (MAMPs and DAMPs), throughput screens of inhibitory molecules. -
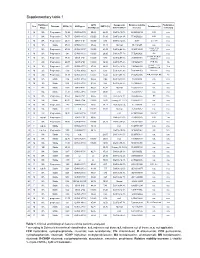
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

ATP-Binding and Hydrolysis in Inflammasome Activation
molecules Review ATP-Binding and Hydrolysis in Inflammasome Activation Christina F. Sandall, Bjoern K. Ziehr and Justin A. MacDonald * Department of Biochemistry & Molecular Biology, Cumming School of Medicine, University of Calgary, 3280 Hospital Drive NW, Calgary, AB T2N 4Z6, Canada; [email protected] (C.F.S.); [email protected] (B.K.Z.) * Correspondence: [email protected]; Tel.: +1-403-210-8433 Academic Editor: Massimo Bertinaria Received: 15 September 2020; Accepted: 3 October 2020; Published: 7 October 2020 Abstract: The prototypical model for NOD-like receptor (NLR) inflammasome assembly includes nucleotide-dependent activation of the NLR downstream of pathogen- or danger-associated molecular pattern (PAMP or DAMP) recognition, followed by nucleation of hetero-oligomeric platforms that lie upstream of inflammatory responses associated with innate immunity. As members of the STAND ATPases, the NLRs are generally thought to share a similar model of ATP-dependent activation and effect. However, recent observations have challenged this paradigm to reveal novel and complex biochemical processes to discern NLRs from other STAND proteins. In this review, we highlight past findings that identify the regulatory importance of conserved ATP-binding and hydrolysis motifs within the nucleotide-binding NACHT domain of NLRs and explore recent breakthroughs that generate connections between NLR protein structure and function. Indeed, newly deposited NLR structures for NLRC4 and NLRP3 have provided unique perspectives on the ATP-dependency of inflammasome activation. Novel molecular dynamic simulations of NLRP3 examined the active site of ADP- and ATP-bound models. The findings support distinctions in nucleotide-binding domain topology with occupancy of ATP or ADP that are in turn disseminated on to the global protein structure. -

Inflammasome Activation and Regulation
Zheng et al. Cell Discovery (2020) 6:36 Cell Discovery https://doi.org/10.1038/s41421-020-0167-x www.nature.com/celldisc REVIEW ARTICLE Open Access Inflammasome activation and regulation: toward a better understanding of complex mechanisms Danping Zheng1,2,TimurLiwinski1,3 and Eran Elinav 1,4 Abstract Inflammasomes are cytoplasmic multiprotein complexes comprising a sensor protein, inflammatory caspases, and in some but not all cases an adapter protein connecting the two. They can be activated by a repertoire of endogenous and exogenous stimuli, leading to enzymatic activation of canonical caspase-1, noncanonical caspase-11 (or the equivalent caspase-4 and caspase-5 in humans) or caspase-8, resulting in secretion of IL-1β and IL-18, as well as apoptotic and pyroptotic cell death. Appropriate inflammasome activation is vital for the host to cope with foreign pathogens or tissue damage, while aberrant inflammasome activation can cause uncontrolled tissue responses that may contribute to various diseases, including autoinflammatory disorders, cardiometabolic diseases, cancer and neurodegenerative diseases. Therefore, it is imperative to maintain a fine balance between inflammasome activation and inhibition, which requires a fine-tuned regulation of inflammasome assembly and effector function. Recently, a growing body of studies have been focusing on delineating the structural and molecular mechanisms underlying the regulation of inflammasome signaling. In the present review, we summarize the most recent advances and remaining challenges in understanding the ordered inflammasome assembly and activation upon sensing of diverse stimuli, as well as the tight regulations of these processes. Furthermore, we review recent progress and challenges in translating inflammasome research into therapeutic tools, aimed at modifying inflammasome-regulated human diseases. -

Functional Screening of ¢Ve PYPAF Family Members Identi¢Es PYPAF5 As a Novel Regulator of NF-UB and Caspase-1
FEBS 26602 FEBS Letters 530 (2002) 73^78 Functional screening of ¢ve PYPAF family members identi¢es PYPAF5 as a novel regulator of NF-UB and caspase-1 Jill M.Grenier 1, Lin Wang1, Gulam A.Manji 2, Waan-Jeng Huang, Amal Al-Garawi, Roxanne Kelly, Adam Carlson, Sarah Merriam, Jose M.Lora, Michael Briskin, Peter S.DiStefano 3, John Bertinà Millennium Pharmaceuticals Inc., 75 Sidney Street, Cambridge, MA 02139, USA Received 22 August 2002; accepted 28 August 2002 First published online 26 September 2002 Edited by Veli-Pekka Lehto activates pro-caspase-9.Apaf-1 has a tripartite domain struc- Abstract PYRIN-containing Apaf-1-like proteins (PYPAFs) are a recently identi¢ed family of proteins thought to function ture consisting of an N-terminal caspase-recruitment domain in apoptotic and in£ammatory signaling pathways. PYPAF1 (CARD) that mediates recruitment of pro-caspase-9 to the and PYPAF7 proteins have been found to assemble with the apoptosome, a central nucleotide-binding site (NBS) domain, PYRIN^CARD protein ASC and coordinate the activation of and a C-terminal domain comprised of WD-40 repeats.The NF-UB and pro-caspase-1. To determine if other PYPAF family NBS domain mediates Apaf-1 oligomerization in the presence members function in pro-in£ammatory signaling pathways, we of dATP, whereas the WD-40 repeats function as binding sites screened ¢ve other PYPAF proteins (PYPAF2, PYPAF3, PY- for cytochrome c.Thus, Apaf-1 functions as a sensor-like PAF4, PYPAF5 and PYPAF6) for their ability to activate NF- molecule that signals apoptosis in response to the release of U B and pro-caspase-1. -

NLR Members in Inflammation-Associated
Cellular & Molecular Immunology (2017) 14, 403–405 & 2017 CSI and USTC All rights reserved 2042-0226/17 $32.00 www.nature.com/cmi RESEARCH HIGHTLIGHT NLR members in inflammation-associated carcinogenesis Ha Zhu1,2 and Xuetao Cao1,2,3 Cellular & Molecular Immunology (2017) 14, 403–405; doi:10.1038/cmi.2017.14; published online 3 April 2017 hronic inflammation is regarded as an impor- nucleotide-binding and oligomerization domain IL-2,8 and NAIP was found to regulate the STAT3 Ctant factor in cancer progression. In addition (NOD)-like receptors (NLRs). TLRs and CLRs are pathway independent of inflammasome formation.9 to the immune surveillance function in the early located in the plasma membranes, whereas RLRs, The AOM/DSS model is the most popular model stage of tumorigenesis, inflammation is also known ALRs and NLRs are intracellular PRRs.3 Unlike used to study the function of NLRs in fl fl as one of the hallmarks of cancer and can supply other families that have been shown to bind their in ammation-associated carcinogenesis. In amma- the tumor microenvironment with bioactive mole- specific cognate ligands, the distinct ligands for somes initiated by NLRs or AIM2 have been widely cules and favor the development of other hallmarks NLRs are still unknown. In fact, mounting evidence reported to participate in the maintenance of 10,11 Nlrp3 Nlrp6 of cancer, such as genetic instability and angiogen- suggests that NLRs function as cytoplasmic sensors intestinal homeostasis. -/-, -/-, Nlrc4 Nlrp1 Nlrx1 Nlrp12 esis. Moreover, inflammation contributes to the and participate in modulating TLR, RLR and CLR -/-, -/-, -/- and -/- mice are 4 more susceptible to AOM/DSS-induced colorectal changing tumor microenvironment by altering signaling pathways. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Apoptosis Ligand-Induced Enhanced Resistance to Fas/Fas
Theileria parva-Transformed T Cells Show Enhanced Resistance to Fas/Fas Ligand-Induced Apoptosis This information is current as Peter Küenzi, Pascal Schneider and Dirk A. E. Dobbelaere of October 2, 2021. J Immunol 2003; 171:1224-1231; ; doi: 10.4049/jimmunol.171.3.1224 http://www.jimmunol.org/content/171/3/1224 Downloaded from References This article cites 69 articles, 29 of which you can access for free at: http://www.jimmunol.org/content/171/3/1224.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on October 2, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2003 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Theileria parva-Transformed T Cells Show Enhanced Resistance to Fas/Fas Ligand-Induced Apoptosis1 Peter Ku¨enzi,2* Pascal Schneider,† and Dirk A. E. Dobbelaere3* Lymphocyte homeostasis is regulated by mechanisms that control lymphocyte proliferation and apoptosis. Activation-induced cell death is mediated by the expression of death ligands and receptors, which, when triggered, activate an apoptotic cascade.