Systemic Effects Downstream of NAIP/NLRC4 Inflammasome Activation in Vivo
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Supplementary Table 2 Supplementary Table 1
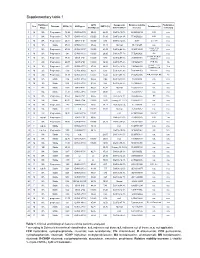
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

ATP-Binding and Hydrolysis in Inflammasome Activation
molecules Review ATP-Binding and Hydrolysis in Inflammasome Activation Christina F. Sandall, Bjoern K. Ziehr and Justin A. MacDonald * Department of Biochemistry & Molecular Biology, Cumming School of Medicine, University of Calgary, 3280 Hospital Drive NW, Calgary, AB T2N 4Z6, Canada; [email protected] (C.F.S.); [email protected] (B.K.Z.) * Correspondence: [email protected]; Tel.: +1-403-210-8433 Academic Editor: Massimo Bertinaria Received: 15 September 2020; Accepted: 3 October 2020; Published: 7 October 2020 Abstract: The prototypical model for NOD-like receptor (NLR) inflammasome assembly includes nucleotide-dependent activation of the NLR downstream of pathogen- or danger-associated molecular pattern (PAMP or DAMP) recognition, followed by nucleation of hetero-oligomeric platforms that lie upstream of inflammatory responses associated with innate immunity. As members of the STAND ATPases, the NLRs are generally thought to share a similar model of ATP-dependent activation and effect. However, recent observations have challenged this paradigm to reveal novel and complex biochemical processes to discern NLRs from other STAND proteins. In this review, we highlight past findings that identify the regulatory importance of conserved ATP-binding and hydrolysis motifs within the nucleotide-binding NACHT domain of NLRs and explore recent breakthroughs that generate connections between NLR protein structure and function. Indeed, newly deposited NLR structures for NLRC4 and NLRP3 have provided unique perspectives on the ATP-dependency of inflammasome activation. Novel molecular dynamic simulations of NLRP3 examined the active site of ADP- and ATP-bound models. The findings support distinctions in nucleotide-binding domain topology with occupancy of ATP or ADP that are in turn disseminated on to the global protein structure. -

NLR Members in Inflammation-Associated
Cellular & Molecular Immunology (2017) 14, 403–405 & 2017 CSI and USTC All rights reserved 2042-0226/17 $32.00 www.nature.com/cmi RESEARCH HIGHTLIGHT NLR members in inflammation-associated carcinogenesis Ha Zhu1,2 and Xuetao Cao1,2,3 Cellular & Molecular Immunology (2017) 14, 403–405; doi:10.1038/cmi.2017.14; published online 3 April 2017 hronic inflammation is regarded as an impor- nucleotide-binding and oligomerization domain IL-2,8 and NAIP was found to regulate the STAT3 Ctant factor in cancer progression. In addition (NOD)-like receptors (NLRs). TLRs and CLRs are pathway independent of inflammasome formation.9 to the immune surveillance function in the early located in the plasma membranes, whereas RLRs, The AOM/DSS model is the most popular model stage of tumorigenesis, inflammation is also known ALRs and NLRs are intracellular PRRs.3 Unlike used to study the function of NLRs in fl fl as one of the hallmarks of cancer and can supply other families that have been shown to bind their in ammation-associated carcinogenesis. In amma- the tumor microenvironment with bioactive mole- specific cognate ligands, the distinct ligands for somes initiated by NLRs or AIM2 have been widely cules and favor the development of other hallmarks NLRs are still unknown. In fact, mounting evidence reported to participate in the maintenance of 10,11 Nlrp3 Nlrp6 of cancer, such as genetic instability and angiogen- suggests that NLRs function as cytoplasmic sensors intestinal homeostasis. -/-, -/-, Nlrc4 Nlrp1 Nlrx1 Nlrp12 esis. Moreover, inflammation contributes to the and participate in modulating TLR, RLR and CLR -/-, -/-, -/- and -/- mice are 4 more susceptible to AOM/DSS-induced colorectal changing tumor microenvironment by altering signaling pathways. -

A Role for the Nlr Family Members Nlrc4 and Nlrp3 in Astrocytic Inflammasome Activation and Astrogliosis
A ROLE FOR THE NLR FAMILY MEMBERS NLRC4 AND NLRP3 IN ASTROCYTIC INFLAMMASOME ACTIVATION AND ASTROGLIOSIS Leslie C. Freeman A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Curriculum of Genetics and Molecular Biology. Chapel Hill 2016 Approved by: Jenny P. Y. Ting Glenn K. Matsushima Beverly H. Koller Silva S. Markovic-Plese Pauline. Kay Lund ©2016 Leslie C. Freeman ALL RIGHTS RESERVED ii ABSTRACT Leslie C. Freeman: A Role for the NLR Family Members NLRC4 and NLRP3 in Astrocytic Inflammasome Activation and Astrogliosis (Under the direction of Jenny P.Y. Ting) The inflammasome is implicated in many inflammatory diseases but has been primarily studied in the macrophage-myeloid lineage. Here we demonstrate a physiologic role for nucleotide-binding domain, leucine-rich repeat, CARD domain containing 4 (NLRC4) in brain astrocytes. NLRC4 has been primarily studied in the context of gram-negative bacteria, where it is required for the maturation of pro-caspase-1 to active caspase-1. We show the heightened expression of NLRC4 protein in astrocytes in a cuprizone model of neuroinflammation and demyelination as well as human multiple sclerotic brains. Similar to macrophages, NLRC4 in astrocytes is required for inflammasome activation by its known agonist, flagellin. However, NLRC4 in astrocytes also mediate inflammasome activation in response to lysophosphatidylcholine (LPC), an inflammatory molecule associated with neurologic disorders. In addition to NLRC4, astrocytic NLRP3 is required for inflammasome activation by LPC. Two biochemical assays show the interaction of NLRC4 with NLRP3, suggesting the possibility of a NLRC4-NLRP3 co-inflammasome. -

AIM2 and NLRC4 Inflammasomes Contribute with ASC to Acute Brain Injury Independently of NLRP3
AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3 Adam Denesa,b,1, Graham Couttsb, Nikolett Lénárta, Sheena M. Cruickshankb, Pablo Pelegrinb,c, Joanne Skinnerb, Nancy Rothwellb, Stuart M. Allanb, and David Broughb,1 aLaboratory of Molecular Neuroendocrinology, Institute of Experimental Medicine, Budapest, 1083, Hungary; bFaculty of Life Sciences, University of Manchester, Manchester M13 9PT, United Kingdom; and cInflammation and Experimental Surgery Unit, CIBERehd (Centro de Investigación Biomédica en Red en el Área temática de Enfermedades Hepáticas y Digestivas), Murcia Biohealth Research Institute–Arrixaca, University Hospital Virgen de la Arrixaca, 30120 Murcia, Spain Edited by Vishva M. Dixit, Genentech, San Francisco, CA, and approved February 19, 2015 (received for review November 18, 2014) Inflammation that contributes to acute cerebrovascular disease is or DAMPs, it recruits ASC, which in turn recruits caspase-1, driven by the proinflammatory cytokine interleukin-1 and is known causing its activation. Caspase-1 then processes pro–IL-1β to a to exacerbate resulting injury. The activity of interleukin-1 is regu- mature form that is rapidly secreted from the cell (5). The ac- lated by multimolecular protein complexes called inflammasomes. tivation of caspase-1 can also cause cell death (6). There are multiple potential inflammasomes activated in diverse A number of inflammasome-forming PRRs have been iden- diseases, yet the nature of the inflammasomes involved in brain tified, including NLR family, pyrin domain containing 1 (NLRP1); injury is currently unknown. Here, using a rodent model of stroke, NLRP3; NLRP6; NLRP7; NLRP12; NLR family, CARD domain we show that the NLRC4 (NLR family, CARD domain containing 4) containing 4 (NLRC4); AIM 2 (absent in melanoma 2); IFI16; and AIM2 (absent in melanoma 2) inflammasomes contribute to and RIG-I (5). -

Apoptosis Ligand-Induced Enhanced Resistance to Fas/Fas
Theileria parva-Transformed T Cells Show Enhanced Resistance to Fas/Fas Ligand-Induced Apoptosis This information is current as Peter Küenzi, Pascal Schneider and Dirk A. E. Dobbelaere of October 2, 2021. J Immunol 2003; 171:1224-1231; ; doi: 10.4049/jimmunol.171.3.1224 http://www.jimmunol.org/content/171/3/1224 Downloaded from References This article cites 69 articles, 29 of which you can access for free at: http://www.jimmunol.org/content/171/3/1224.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on October 2, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2003 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Theileria parva-Transformed T Cells Show Enhanced Resistance to Fas/Fas Ligand-Induced Apoptosis1 Peter Ku¨enzi,2* Pascal Schneider,† and Dirk A. E. Dobbelaere3* Lymphocyte homeostasis is regulated by mechanisms that control lymphocyte proliferation and apoptosis. Activation-induced cell death is mediated by the expression of death ligands and receptors, which, when triggered, activate an apoptotic cascade. -

NAIP5/NLRC4 Inflammasomes Compounds Inhibit the NLRP1, NLRP3, and Arsenic Trioxide and Other Arsenical
The Journal of Immunology Arsenic Trioxide and Other Arsenical Compounds Inhibit the NLRP1, NLRP3, and NAIP5/NLRC4 Inflammasomes Nolan K. Maier,* Devorah Crown,* Jie Liu,† Stephen H. Leppla,* and Mahtab Moayeri* Inflammasomes are large cytoplasmic multiprotein complexes that activate caspase-1 in response to diverse intracellular danger signals. Inflammasome components termed nucleotide-binding oligomerization domain–like receptor (NLR) proteins act as sensors for pathogen-associated molecular patterns, stress, or danger stimuli. We discovered that arsenicals, including arsenic trioxide and sodium arsenite, inhibited activation of the NLRP1, NLRP3, and NAIP5/NLRC4 inflammasomes by their respective activat- ing signals, anthrax lethal toxin, nigericin, and flagellin. These compounds prevented the autoproteolytic activation of caspase-1 and the processing and secretion of IL-1b from macrophages. Inhibition was independent of protein synthesis induction, proteasome-mediated protein breakdown, or kinase signaling pathways. Arsenic trioxide and sodium arsenite did not directly modify or inhibit the activity of preactivated recombinant caspase-1. Rather, they induced a cellular state inhibitory to both the autoproteolytic and substrate cleavage activities of caspase-1, which was reversed by the reactive oxygen species scavenger N-acetylcysteine but not by reducing agents or NO pathway inhibitors. Arsenicals provided protection against NLRP1-dependent anthrax lethal toxin–mediated cell death and prevented NLRP3-dependent neutrophil recruitment in a monosodium urate crystal inflammatory murine peritonitis model. These findings suggest a novel role in inhibition of the innate immune response for arsenical compounds that have been used as therapeutics for a few hundred years. The Journal of Immunology, 2014, 192: 763–770. nflammasomes are large cytoplasmic multiprotein complexes domain–containing protein 4 (NLRC4) inflammasome by direct that form in response to intracellular danger signals. -

Techniques for Immune Function Analysis Application Handbook 1St Edition
Techniques for Immune Function Analysis Application Handbook 1st Edition BD Biosciences For additional information please access the Immune Function Homepage at www.bdbiosciences.com/immune_function For Research Use Only. Not for use in diagnostic or therapeutic procedures. Purchase does not include or carry any right to resell or transfer this product either as a stand-alone product or as a component of another product. Any use of this product other than the permitted use without the express written authorization of Becton Dickinson and Company is strictly prohibited. All applications are either tested in-house or reported in the literature. See Technical Data Sheets for details. BD, BD Logo and all other trademarks are the property of Becton, Dickinson and Company. ©2003 BD Table of Contents Preface . 4 Chapter 1: Immunofluorescent Staining of Cell Surface Molecules for Flow Cytometric Analysis . 9 Chapter 2: BD™ Cytometric Bead Array (CBA) Multiplexing Assays . 35 Chapter 3: BD™ DimerX MHC:Ig Proteins for the Analysis of Antigen-specific T Cells. 51 Chapter 4: Immunofluorescent Staining of Intracellular Molecules for Flow Cytometric Analysis . 61 Chapter 5: BD FastImmune™ Cytokine Flow Cytometry. 85 Chapter 6: BD™ ELISPOT Assays for Cells That Secrete Biological Response Modifiers . 109 Chapter 7: ELISA for Specifically Measuring the Levels of Cytokines, Chemokines, Inflammatory Mediators and their Receptors . 125 Chapter 8: BD OptEIA™ ELISA Sets and Kits for Quantitation of Analytes in Serum, Plasma, and Cell Culture Supernatants. 143 Chapter 9: BrdU Staining and Multiparameter Flow Cytometric Analysis of the Cell Cycle . 155 Chapter 10: Cell-based Assays for Biological Response Modifiers . 177 Chapter 11: BD RiboQuant™ Multi-Probe RNase Protection Assay System . -

Molecular Genetics of Spinal Muscular Atrophy: Contribution of the NAIP Gene to Clinical Severity
Kobe J. Med. Sci. 48, 25/31 February 2002 Molecular Genetics of Spinal Muscular Atrophy: Contribution of the NAIP Gene to Clinical Severity TOMOKO AKUTSU1*, HISAHIDE NISHIO2, KIMIAKI SUMINO2, YASUHIRO TAKESHIMA1, SYUICHI TSUNEISHI1, HIROKO WADA1, SATOSHI TAKADA3, MASAFUMI MATSUO1, 4, and HAJIME NAKAMURA1 Division of Pediatrics, Department of Development and Aging1, Division of Public Health, Department of Environmental Health and Safety2, Kobe University Graduate School of Medicine Faculty of Health Science, Kobe University School of Medicine, Kobe 654-0142, Japan3 Division of Molecular Medicine, Department of International and Environmental Medical Sciences, Kobe University Graduate School of Medicine4 Received 5 February 2002/ Accepted 19 February 2002 Key words: spinal muscular atrophy; the SMN1 gene; the NAIP gene; the p44t gene Spinal muscular atrophy (SMA) is one of the most common autosomal recessive disorders characterized by degeneration of anterior horn cells in the spinal cord, and leads to progressive muscular weakness and atrophy. At least three SMA-related genes have been identified: SMN1, NAIP and p44t. We analyzed these genes in 32 SMA patients and found that the SMN1 gene was deleted in 30 of 32 patients (94 %), irrespective of clinical type. The NAIP gene was deleted in 6 patients and its deletion rate was higher in type I patients than that in typeⅡ or Ⅲ. Further, in type I patients lacking the NAIP gene, deterioration in their respiratory function is more rapid than in those type I patients retaining the NAIP gene. Since complete p44t deletion was observed in only 3 patients, the correlation between the p44t deletion and severity of SMA remained ambiguous. -

Evasion of Inflammasome Activation by Microbial Pathogens
Evasion of inflammasome activation by microbial pathogens Tyler K. Ulland, … , Polly J. Ferguson, Fayyaz S. Sutterwala J Clin Invest. 2015;125(2):469-477. https://doi.org/10.1172/JCI75254. Review Activation of the inflammasome occurs in response to infection with a wide array of pathogenic microbes. The inflammasome serves as a platform to activate caspase-1, which results in the subsequent processing and secretion of the proinflammatory cytokines IL-1β and IL-18 and the initiation of an inflammatory cell death pathway termed pyroptosis. Effective inflammasome activation is essential in controlling pathogen replication as well as initiating adaptive immune responses against the offending pathogens. However, a number of pathogens have developed strategies to evade inflammasome activation. In this Review, we discuss these pathogen evasion strategies as well as the potential infectious complications of therapeutic blockade of IL-1 pathways. Find the latest version: https://jci.me/75254/pdf The Journal of Clinical Investigation REVIEW Evasion of inflammasome activation by microbial pathogens Tyler K. Ulland,1,2 Polly J. Ferguson,3 and Fayyaz S. Sutterwala1,2,4,5 1Inflammation Program, 2Interdisciplinary Program in Molecular and Cellular Biology, 3Department of Pediatrics, and 4Department of Internal Medicine, University of Iowa, Iowa City, Iowa, USA. 5Veterans Affairs Medical Center, Iowa City, Iowa, USA. Activation of the inflammasome occurs in response to infection with a wide array of pathogenic microbes. The inflammasome serves as a platform to activate caspase-1, which results in the subsequent processing and secretion of the proinflammatory cytokines IL-1β and IL-18 and the initiation of an inflammatory cell death pathway termed pyroptosis. -

The Role of Nucleolin in B-Cell Lymphomas and Fas-Mediated Apoptotic Signaling
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 5-2013 The Role of Nucleolin in B-cell Lymphomas and Fas-Mediated Apoptotic Signaling Jillian F. Wise Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Cancer Biology Commons, and the Medicine and Health Sciences Commons Recommended Citation Wise, Jillian F., "The Role of Nucleolin in B-cell Lymphomas and Fas-Mediated Apoptotic Signaling" (2013). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 339. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/339 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. The Role of Nucleolin in B-cell Lymphomas and Fas-Mediated Apoptotic Signaling by Jillian F Wise, BS Approved: ___________________________________________________ Felipe -

NOD-Like Receptors (Nlrs) and Inflammasomes
International Edition www.adipogen.com NOD-like Receptors (NLRs) and Inflammasomes In mammals, germ-line encoded pattern recognition receptors (PRRs) detect the presence of pathogens through recognition of pathogen-associated molecular patterns (PAMPs) or endogenous danger signals through the sensing of danger-associated molecular patterns (DAMPs). The innate immune system comprises several classes of PRRs that allow the early detection of pathogens at the site of infection. The membrane-bound toll-like receptors (TLRs) and C-type lectin receptors (CTRs) detect PAMPs in extracellular milieu and endo- somal compartments. TRLs and CTRs cooperate with PRRs sensing the presence of cytosolic nucleic acids, like RNA-sensing RIG-I (retinoic acid-inducible gene I)-like receptors (RLRs; RLHs) or DNA-sensing AIM2, among others. Another set of intracellular sensing PRRs are the NOD-like receptors (NLRs; nucleotide-binding domain leucine-rich repeat containing receptors), which not only recognize PAMPs but also DAMPs. PAMPs FUNGI/PROTOZOA BACTERIA VIRUSES MOLECULES C. albicans A. hydrophila Adenovirus Bacillus anthracis lethal Plasmodium B. brevis Encephalomyo- toxin (LeTx) S. cerevisiae E. coli carditis virus Bacterial pore-forming L. monocytogenes Herpes simplex virus toxins L. pneumophila Influenza virus Cytosolic dsDNA N. gonorrhoeae Sendai virus P. aeruginosa Cytosolic flagellin S. aureus MDP S. flexneri meso-DAP S. hygroscopicus S. typhimurium DAMPs MOLECULES PARTICLES OTHERS DNA Uric acid UVB Extracellular ATP CPPD Mutations R837 Asbestos Cytosolic dsDNA Irritants Silica Glucose Alum Hyaluronan Amyloid-b Hemozoin Nanoparticles FIGURE 1: Overview on PAMPs and DAMPs recognized by NLRs. NOD-like Receptors [NLRs] The intracellular NLRs organize signaling complexes such as inflammasomes and NOD signalosomes.