The Roles of Various Prostaglandins in Fibrosis: a Review
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) United States Patent (10) Patent No.: US 9,498,481 B2 Rao Et Al
USOO9498481 B2 (12) United States Patent (10) Patent No.: US 9,498,481 B2 Rao et al. (45) Date of Patent: *Nov. 22, 2016 (54) CYCLOPROPYL MODULATORS OF P2Y12 WO WO95/26325 10, 1995 RECEPTOR WO WO99/O5142 2, 1999 WO WOOO/34283 6, 2000 WO WO O1/92262 12/2001 (71) Applicant: Apharaceuticals. Inc., La WO WO O1/922.63 12/2001 olla, CA (US) WO WO 2011/O17108 2, 2011 (72) Inventors: Tadimeti Rao, San Diego, CA (US); Chengzhi Zhang, San Diego, CA (US) OTHER PUBLICATIONS Drugs of the Future 32(10), 845-853 (2007).* (73) Assignee: Auspex Pharmaceuticals, Inc., LaJolla, Tantry et al. in Expert Opin. Invest. Drugs (2007) 16(2):225-229.* CA (US) Wallentin et al. in the New England Journal of Medicine, 361 (11), 1045-1057 (2009).* (*) Notice: Subject to any disclaimer, the term of this Husted et al. in The European Heart Journal 27, 1038-1047 (2006).* patent is extended or adjusted under 35 Auspex in www.businesswire.com/news/home/20081023005201/ U.S.C. 154(b) by Od en/Auspex-Pharmaceuticals-Announces-Positive-Results-Clinical M YW- (b) by ayS. Study (published: Oct. 23, 2008).* This patent is Subject to a terminal dis- Concert In www.concertpharma. com/news/ claimer ConcertPresentsPreclinicalResultsNAMS.htm (published: Sep. 25. 2008).* Concert2 in Expert Rev. Anti Infect. Ther. 6(6), 782 (2008).* (21) Appl. No.: 14/977,056 Springthorpe et al. in Bioorganic & Medicinal Chemistry Letters 17. 6013-6018 (2007).* (22) Filed: Dec. 21, 2015 Leis et al. in Current Organic Chemistry 2, 131-144 (1998).* Angiolillo et al., Pharmacology of emerging novel platelet inhibi (65) Prior Publication Data tors, American Heart Journal, 2008, 156(2) Supp. -

Prevention of Atherothrombotic Events in Patients with Diabetes Mellitus: from Antithrombotic Therapies to New-Generation Glucose-Lowering Drugs
CONSENSUS STATEMENT EXPERT CONSENSUS DOCUMENT Prevention of atherothrombotic events in patients with diabetes mellitus: from antithrombotic therapies to new-generation glucose-lowering drugs Giuseppe Patti1*, Ilaria Cavallari2, Felicita Andreotti3, Paolo Calabrò4, Plinio Cirillo5, Gentian Denas6, Mattia Galli3, Enrica Golia4, Ernesto Maddaloni 7, Rossella Marcucci8, Vito Maurizio Parato9,10, Vittorio Pengo6, Domenico Prisco8, Elisabetta Ricottini2, Giulia Renda11, Francesca Santilli12, Paola Simeone12 and Raffaele De Caterina11,13*, on behalf of the Working Group on Thrombosis of the Italian Society of Cardiology Abstract | Diabetes mellitus is an important risk factor for a first cardiovascular event and for worse outcomes after a cardiovascular event has occurred. This situation might be caused, at least in part, by the prothrombotic status observed in patients with diabetes. Therefore, contemporary antithrombotic strategies, including more potent agents or drug combinations, might provide greater clinical benefit in patients with diabetes than in those without diabetes. In this Consensus Statement, our Working Group explores the mechanisms of platelet and coagulation activity , the current debate on antiplatelet therapy in primary cardiovascular disease prevention, and the benefit of various antithrombotic approaches in secondary prevention of cardiovascular disease in patients with diabetes. While acknowledging that current data are often derived from underpowered, observational studies or subgroup analyses of larger trials, we propose -

Xử Trí Quá Liều Thuốc Chống Đông
XỬ TRÍ QUÁ LIỀU THUỐC CHỐNG ĐÔNG TS. Trần thị Kiều My Bộ môn Huyết học Đại học Y Hà nội Khoa Huyết học Bệnh viện Bạch mai TS. Trần thị Kiều My Coagulation cascade TIÊU SỢI HUYẾT THUỐC CHỐNG ĐÔNG VÀ CHỐNG HUYẾT KHỐI • Thuốc chống ngưng tập tiểu cầu • Thuốc chống các yếu tố đông máu huyết tương • Thuốc tiêu sợi huyết và huyết khối Thuốc chống ngưng tập tiểu cầu Đường sử Xét nghiệm theo dõi dụng Nhóm thuốc ức chế Glycoprotein IIb/IIIa: TM VerifyNow Abciximab, ROTEM Tirofiban SLTC, Hb, Hct, APTT, Clotting time Eptifibatide, ACT, aPTT, TT, and PT Nhóm ức chế receptor ADP /P2Y12 Uống NTTC, phân tích chức +thienopyridines năng TC bằng PFA, Clopidogrel ROTEM Prasugrel VerifyNow Ticlopidine +nucleotide/nucleoside analogs Cangrelor Elinogrel, TM Ticagrelor TM+uống ƯC ADP Uống Nhóm Prostaglandin analogue (PGI2): Uống NTTC, phân tích chức Beraprost, năng TC bằng PFA, ROTEM Iloprost (Illomedin), Xịt hoặc truyền VerifyNow TM Prostacyclin,Treprostinil Nhóm ức chế COX: Uống NTTC, phân tích Acetylsalicylicacid/Aspirin#Aloxiprin,Carbasalate, chức năng TC bằng calcium, Indobufen, Triflusal PFA, ROTEM VerifyNow Nhóm ức chế Thromboxane: Uống NTTC, phân tích chức +thromboxane synthase inhibitors năng TC bằng PFA, Dipyridamole (+Aspirin), Picotamide ROTEM +receptor antagonist : Terutroban† VerifyNow Nhóm ức chế Phosphodiesterase: Uống NTTC, phân tích chức Cilostazol, Dipyridamole, Triflusal năng TC bằng PFA, ROTEM VerifyNow Nhóm khác: Uống NTTC, phân tích chức Cloricromen, Ditazole, Vorapaxar năng TC bằng PFA, ROTEM VerifyNow Dược động học một số thuốc -

Estimation of Serum Prostaglandin D2 Levels and Its Expression in Tissue of Alopecia Areata
ISSN: 2536-9474 (Print) Original article / FYMJ ISSN: 2536-9482 (Online) Fayoum University Medical Journal Soliman et al., 2019,4(1), 77-85 Estimation of serum prostaglandin D2 levels and its expression in tissue of Alopecia areata Talal A. Abd-ElRaheem1, Samar M.R. El-Tahlawy2, Olfat G. Shaker3, Mohamed H.Mohamed4 and Yasmin F.Soliman5 1. M.D, professor of Dermatology, STDs and Andrology department, Faculty of Medicine Fayoum University. 2. M.D, professor of Dermatology department, Faculty of Medicine Cairo University 3. M.D, professor of Biochemistry department, Faculty of Medicine-Cairo University 4. MD, lecturer of Dermatology, STDs and Andrology department, Faculty of Medicine Fayoum University 5. M.B.B.CH, Dermatology, STDs and Andrology department, Faculty of Medicine, Cairo University Abstract Results: There was statistically highly Back ground: Alopecia areata is a recurrent, significant difference between the two groups non-scaring type of hair loss considered to be regarding the mean value of PGD2 in tissue in an autoimmune process. Though its AA patients. It was significantly lower than in etiopathogenesis is not fully understood, many control group (p < 0.001). The mean value of therapeutic options have been used by PGD2 in serum in AA patients was dermatologists, but none are curative or significantly lower than in control group (p< preventive. Prostaglandins analogues which 0.05). are used to treat glaucoma. Increase in eye lash Conclusion: Prostaglandin D2 exhibits a number, thickness and pigmentation have been strong role in etiology of alopecia areata and reported as side effect. significantly was elevated in serum and tissue Methods: This cross sectional case control of alopecia areata patients. -

Antithrombotic Treatment After Stroke Due to Intracerebral Haemorrhage (Review)
Cochrane Database of Systematic Reviews Antithrombotic treatment after stroke due to intracerebral haemorrhage (Review) Perry LA, Berge E, Bowditch J, Forfang E, Rønning OM, Hankey GJ, Villanueva E, Al-Shahi Salman R Perry LA, Berge E, Bowditch J, Forfang E, Rønning OM, Hankey GJ, Villanueva E, Al-Shahi Salman R. Antithrombotic treatment after stroke due to intracerebral haemorrhage. Cochrane Database of Systematic Reviews 2017, Issue 5. Art. No.: CD012144. DOI: 10.1002/14651858.CD012144.pub2. www.cochranelibrary.com Antithrombotic treatment after stroke due to intracerebral haemorrhage (Review) Copyright © 2017 The Cochrane Collaboration. Published by John Wiley & Sons, Ltd. TABLE OF CONTENTS HEADER....................................... 1 ABSTRACT ...................................... 1 PLAINLANGUAGESUMMARY . 2 SUMMARY OF FINDINGS FOR THE MAIN COMPARISON . ..... 3 BACKGROUND .................................... 5 OBJECTIVES ..................................... 5 METHODS ...................................... 6 RESULTS....................................... 8 Figure1. ..................................... 9 Figure2. ..................................... 11 Figure3. ..................................... 12 DISCUSSION ..................................... 14 AUTHORS’CONCLUSIONS . 15 ACKNOWLEDGEMENTS . 15 REFERENCES ..................................... 15 CHARACTERISTICSOFSTUDIES . 18 DATAANDANALYSES. 31 Analysis 1.2. Comparison 1 Short-term antithrombotic treatment, Outcome 2 Death. 31 Analysis 1.6. Comparison 1 Short-term antithrombotic -
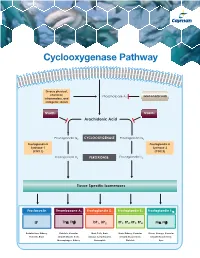
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

New Investigational Drugs for Androgenetic Alopecia. Valente Duarte De Sousa IC 1, Tosti A
Expert Opin Investig Drugs. 2013 May;22(5):573-89. doi: 10.1517/13543784.2013.784743. Epub 2013 Apr 4. New investigational drugs for androgenetic alopecia. Valente Duarte de Sousa IC 1, Tosti A . Author information • [email protected] Erratum in • Erratum. [Expert Opin Investig Drugs. 2015] Abstract INTRODUCTION: Androgenetic alopecia (AGA) is the most common form of hair loss, however current treatment options are limited and moderately effective. In the past few years, there has been an increased interest in deciphering the molecular mechanisms responsible for this disorder, which has opened the possibility of novel treatments that promise to not only stimulate hair growth, but also to induce formation of new hair follicles. AREAS COVERED: The future holds more effective topical treatments with less systemic side effects (such as topical 5- alfa-reductase inhibitors), prostaglandin analogs and antagonists, medications which act through the Wnt signaling pathway, stem cells for hair regeneration, platelet-rich plasma (PRP) and more effective ways of transplanting hair. A comprehensive search was made using PubMed, GoogleScholar and Clinicaltrial.gov using different combination of key words, which included AGA treatment, new treatments for AGA, Wnt pathway, prostaglandins, PRP and stem cells for hair regrowth. EXPERT OPINION: In the near future, treatments with topical 5-alfa-reductase inhibitors and prostaglandin agonists or antagonists are expected. More evidence is needed to verify the efficacy of PRP. Although hair follicle bioengineering and multiplication is a fascinating and promising field, it is still a long way from being available to clinicians. J Am Acad Dermatol. 2015 Apr;72(4):712-6. -

Vasoactive Responses of U46619, PGF2 , Latanoprost, and Travoprost
Vasoactive Responses of U46619, PGF2␣, Latanoprost, and Travoprost in Isolated Porcine Ciliary Arteries Ineta Vysniauskiene, Reto Allemann, Josef Flammer, and Ivan O. Haefliger PURPOSE. To compare the vasoactive properties of the prosta- these responses can be modulated by SQ 29548 (TP-receptor noids U46619 (thromboxane A2 analogue), prostaglandin F2␣ antagonist) or AL-8810 (FP-receptor antagonist). (PGF2␣), latanoprost free acid, and travoprost free acid in isolated porcine ciliary arteries. METHODS. In a myograph system (isometric force measure- MATERIAL AND METHODS ment), quiescent vessels were exposed (cumulatively) to U46619, PGF , latanoprost, or travoprost (0.1 nM–0.1 mM). 2␣ Vessels Preparation Experiments were also conducted in the presence of SQ 29548 (TP-receptor antagonist; 3–10 M) or AL-8810 (FP-receptor Porcine eyes were obtained from an abattoir immediately after death. antagonist; 3–30 M). Contractions were expressed as the In cold modified Krebs-Ringer solution (NaCl 118 mM, KCl 4.7 mM, percentage of 100 mM potassium chloride-induced contrac- CaCl2 2.5 mM, K2PO4 1.2 mM, MgSO4 1.2 mM, NaHCO3 25 mM, tions. glucose 11.1 mM, and EDTA 0.026 mM), ciliary arteries were dissected 5 RESULTS. In quiescent vessels, contractions (concentration–re- and cut into 2-mm segments. In an organ chamber, two 45- m Ϯ tungsten wires were passed through the vessel’s lumen and attached to sponse curves) induced by (0.1 mM) PGF2␣ (87.9% 3.5%), U46619 (66.7% Ϯ 4.1%), and latanoprost (62.9% Ϯ 3.6%) were a force transducer for isometric force measurements (Myo-Interface; JP more pronounced (P Յ 0.001) than those induced by tra- Trading, Aarhus, Denmark). -

Clinical Trial Protocol: BPS-314D-MR-PAH-302
Clinical Trial Protocol: BPS-314d-MR-PAH-302 Study Title: A multicenter, double-blind, randomized, placebo-controlled, Phase 3 study to assess the efficacy and safety of oral BPS-314d-MR added-on to treprostinil, inhaled (Tyvaso®) in subjects with pulmonary arterial hypertension Study Number: BPS-314d- MR-PAH-302 Study Phase: 3 Product Name: Beraprost Sodium 314d Modified Release IND Number: 111,729 Indication: Treatment of Pulmonary Arterial Hypertension Investigators: Multicenter Sponsor: Lung Biotechnology Inc. Sponsor Contact: Medical Monitor: Date Original Protocol: 16 April 2013 Amendment 1: 17 December 2013 Amendment 2 15 October 2014 GCP Statement: This study will be conducted in compliance with the protocol, Good Clinical Practice (GCP) and applicable regulatory requirements. Confidentiality Statement The concepts and information contained herein are confidential and proprietary and shall not be disclosed in whole or part without the express written consent of Lung Biotechnology Inc. © 2014 Lung Biotechnology Inc. Beraprost Sodium 314d Modified Release Lung Biotechnology Inc. Clinical Trial Protocol: BPS-314-d-MR-PAH 302 15 October 2014 LIST OF CONTACTS Study Sponsor: Lung Biotechnology Inc. 1040 Spring Street Silver Spring, Maryland 20910 United States Phone: 301-608-9292 Fax: 301-589-0855 Sponsor Contact: Medical Monitor: SAE Reporting: CONFIDENTIAL Page 2 of 75 Beraprost Sodium 314d Modified Release Lung Biotechnology Inc. Clinical Trial Protocol: BPS-314-d-MR-PAH 302 15 October 2014 SYNOPSIS Sponsor: Lung Biotechnology Inc. -

Effect of Prostanoids on Human Platelet Function: an Overview
International Journal of Molecular Sciences Review Effect of Prostanoids on Human Platelet Function: An Overview Steffen Braune, Jan-Heiner Küpper and Friedrich Jung * Institute of Biotechnology, Molecular Cell Biology, Brandenburg University of Technology, 01968 Senftenberg, Germany; steff[email protected] (S.B.); [email protected] (J.-H.K.) * Correspondence: [email protected] Received: 23 October 2020; Accepted: 23 November 2020; Published: 27 November 2020 Abstract: Prostanoids are bioactive lipid mediators and take part in many physiological and pathophysiological processes in practically every organ, tissue and cell, including the vascular, renal, gastrointestinal and reproductive systems. In this review, we focus on their influence on platelets, which are key elements in thrombosis and hemostasis. The function of platelets is influenced by mediators in the blood and the vascular wall. Activated platelets aggregate and release bioactive substances, thereby activating further neighbored platelets, which finally can lead to the formation of thrombi. Prostanoids regulate the function of blood platelets by both activating or inhibiting and so are involved in hemostasis. Each prostanoid has a unique activity profile and, thus, a specific profile of action. This article reviews the effects of the following prostanoids: prostaglandin-D2 (PGD2), prostaglandin-E1, -E2 and E3 (PGE1, PGE2, PGE3), prostaglandin F2α (PGF2α), prostacyclin (PGI2) and thromboxane-A2 (TXA2) on platelet activation and aggregation via their respective receptors. Keywords: prostacyclin; thromboxane; prostaglandin; platelets 1. Introduction Hemostasis is a complex process that requires the interplay of multiple physiological pathways. Cellular and molecular mechanisms interact to stop bleedings of injured blood vessels or to seal denuded sub-endothelium with localized clot formation (Figure1). -

Activation of the Murine EP3 Receptor for PGE2 Inhibits Camp Production and Promotes Platelet Aggregation
Activation of the murine EP3 receptor for PGE2 inhibits cAMP production and promotes platelet aggregation Jean-Etienne Fabre, … , Thomas M. Coffman, Beverly H. Koller J Clin Invest. 2001;107(5):603-610. https://doi.org/10.1172/JCI10881. Article The importance of arachidonic acid metabolites (termed eicosanoids), particularly those derived from the COX-1 and COX-2 pathways (termed prostanoids), in platelet homeostasis has long been recognized. Thromboxane is a potent agonist, whereas prostacyclin is an inhibitor of platelet aggregation. In contrast, the effect of prostaglandin E2 (PGE2) on platelet aggregation varies significantly depending on its concentration. Low concentrations of PGE2 enhance platelet aggregation, whereas high PGE2 levels inhibit aggregation. The mechanism for this dual action of PGE2 is not clear. This study shows that among the four PGE2 receptors (EP1–EP4), activation of EP3 is sufficient to mediate the proaggregatory actions of low PGE2 concentration. In contrast, the prostacyclin receptor (IP) mediates the inhibitory effect of higher PGE2 concentrations. Furthermore, the relative activation of these two receptors, EP3 and IP, regulates the intracellular level of cAMP and in this way conditions the response of the platelet to aggregating agents. Consistent with these findings, loss of the EP3 receptor in a model of venous inflammation protects against formation of intravascular clots. Our results suggest that local production of PGE2 during an inflammatory process can modulate ensuing platelet responses. Find the latest version: https://jci.me/10881/pdf Activation of the murine EP3 receptor for PGE2 inhibits cAMP production and promotes platelet aggregation Jean-Etienne Fabre,1 MyTrang Nguyen,1 Krairek Athirakul,2 Kenneth Coggins,1 John D. -

Antiplatelet Effects of Prostacyclin Analogues: Which One to Choose in Case of Thrombosis Or Bleeding? Sylwester P
INTERVENTIONAL CARDIOLOGY Cardiology Journal 20XX, Vol. XX, No. X, XXX–XXX DOI: 10.5603/CJ.a2020.0164 Copyright © 20XX Via Medica REVIEW ARTICLE ISSN 1897–5593 eISSN 1898–018X Antiplatelet effects of prostacyclin analogues: Which one to choose in case of thrombosis or bleeding? Sylwester P. Rogula1*, Hubert M. Mutwil1*, Aleksandra Gąsecka1, Marcin Kurzyna2, Krzysztof J. Filipiak1 11st Chair and Department of Cardiology, Medical University of Warsaw, Poland 2Department of Pulmonary Circulation, Thromboembolic Diseases and Cardiology, Center of Postgraduate Education Medical, European Health Center Otwock, Poland Abstract Prostacyclin and analogues are successfully used in the treatment of pulmonary arterial hypertension (PAH) due to their vasodilatory effect on pulmonary arteries. Besides vasodilatory effect, prostacyclin analogues inhibit platelets, but their antiplatelet effect is not thoroughly established. The antiplatelet effect of prostacyclin analogues may be beneficial in case of increased risk of thromboembolic events, or undesirable in case of increased risk of bleeding. Since prostacyclin and analogues differ regarding their potency and form of administration, they might also inhibit platelets to a different extent. This review summarizes the recent evidence on the antiplatelet effects of prostacyclin and analogue in the treatment of PAH, this is important to consider when choosing the optimal treatment regimen in tailoring to an individual patients’ needs. (Cardiol J 20XX; XX, X: xx–xx) Key words: prostacyclin analogues, pulmonary arterial hypertension, platelets, antiplatelet effect, thrombosis, bleeding Introduction tiproliferative effects [4]. The main indication for PGI2 and analogues is advanced pulmonary arterial Since 1935 when prostaglandin was isolated hypertension (PAH) and peripheral vascular disor- for the first time [1], many scientists have focused ders [5].