Comparative Analysis of Camelid Mitochondrial Genomes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Redalyc.Bienestar Animal En Bovinos De Leche: Selección De Indicadores
RIA. Revista de Investigaciones Agropecuarias ISSN: 0325-8718 [email protected] Instituto Nacional de Tecnología Agropecuaria Argentina MARTÍNEZ, G. M.; SUÁREZ, V. H.; GHEZZI, M. D. Bienestar animal en bovinos de leche: selección de indicadores vinculados a la salud y producción RIA. Revista de Investigaciones Agropecuarias, vol. 42, núm. 2, agosto, 2016, pp. 153- 160 Instituto Nacional de Tecnología Agropecuaria Buenos Aires, Argentina Disponible en: http://www.redalyc.org/articulo.oa?id=86447075008 Cómo citar el artículo Número completo Sistema de Información Científica Más información del artículo Red de Revistas Científicas de América Latina, el Caribe, España y Portugal Página de la revista en redalyc.org Proyecto académico sin fines de lucro, desarrollado bajo la iniciativa de acceso abierto Agosto 2016, Argentina 15 Bienestar animal en bovinos de leche: selección de indicadores vinculados a la salud y producción MARTÍNEZ, G. M. 1*; SUÁREZ, V. H. 2; GHEZZI, M. D. 3 RESUMEN El bienestar animal ha sido denido por la Organización Mundial de Sanidad Animal (OIE) como el término amplio que describe la manera en que los individuos se enfrentan con el ambiente y que incluye su sanidad, sus percepciones, su estado anímico y otros e fectos positivos o negativos que inuyen sobre los mecanis - mos físicos y psíquicos del animal. Durante años se tuvo como objetivo principal dentro de los programas de mejora genética en rodeos lecheros el aumento en la producción de leche por individuo; posteriormente se trabajó en compatibilizar ese incremento en el rinde con una mayor eciencia en la conversión alimenticia. A lo largo de este período todo el sistema productivo se fue transformando de manera tal de ofrecerle a esos animales de alto mérito genético el ambiente necesario para que consiguiesen expresar su potencial. -

(Lama Guanicoe) in South America
Molecular Ecology (2013) 22, 463–482 doi: 10.1111/mec.12111 The influence of the arid Andean high plateau on the phylogeography and population genetics of guanaco (Lama guanicoe) in South America JUAN C. MARIN,* BENITO A. GONZA´ LEZ,† ELIE POULIN,‡ CIARA S. CASEY§ and WARREN E. JOHNSON¶ *Laboratorio de Geno´mica y Biodiversidad, Departamento de Ciencias Ba´sicas, Facultad de Ciencias, Universidad del Bı´o-Bı´o, Casilla 447, Chilla´n, Chile, †Laboratorio de Ecologı´a de Vida Silvestre, Facultad de Ciencias Forestales y de la Conservacio´ndela Naturaleza, Universidad de Chile, Santiago, Chile, ‡Laboratorio de Ecologı´a Molecular, Departamento de Ciencias Ecolo´gicas, Facultad de Ciencias, Instituto de Ecologı´a y Biodiversidad, Universidad de Chile, Santiago, Chile, §Department of Biological Science, University of Lincoln, Lincoln, UK, ¶Laboratory of Genomic Diversity, National Cancer Institute, Frederick, MD, USA Abstract A comprehensive study of the phylogeography and population genetics of the largest wild artiodactyl in the arid and cold-temperate South American environments, the gua- naco (Lama guanicoe) was conducted. Patterns of molecular genetic structure were described using 514 bp of mtDNA sequence and 14 biparentally inherited microsatel- lite markers from 314 samples. These individuals originated from 17 localities through- out the current distribution across Peru, Bolivia, Argentina and Chile. This confirmed well-defined genetic differentiation and subspecies designation of populations geo- graphically separated to the northwest (L. g. cacsilensis) and southeast (L. g. guanicoe) of the central Andes plateau. However, these populations are not completely isolated, as shown by admixture prevalent throughout a limited contact zone, and a strong sig- nal of expansion from north to south in the beginning of the Holocene. -

Prospects for Rewilding with Camelids
Journal of Arid Environments 130 (2016) 54e61 Contents lists available at ScienceDirect Journal of Arid Environments journal homepage: www.elsevier.com/locate/jaridenv Prospects for rewilding with camelids Meredith Root-Bernstein a, b, *, Jens-Christian Svenning a a Section for Ecoinformatics & Biodiversity, Department of Bioscience, Aarhus University, Aarhus, Denmark b Institute for Ecology and Biodiversity, Santiago, Chile article info abstract Article history: The wild camelids wild Bactrian camel (Camelus ferus), guanaco (Lama guanicoe), and vicuna~ (Vicugna Received 12 August 2015 vicugna) as well as their domestic relatives llama (Lama glama), alpaca (Vicugna pacos), dromedary Received in revised form (Camelus dromedarius) and domestic Bactrian camel (Camelus bactrianus) may be good candidates for 20 November 2015 rewilding, either as proxy species for extinct camelids or other herbivores, or as reintroductions to their Accepted 23 March 2016 former ranges. Camels were among the first species recommended for Pleistocene rewilding. Camelids have been abundant and widely distributed since the mid-Cenozoic and were among the first species recommended for Pleistocene rewilding. They show a range of adaptations to dry and marginal habitats, keywords: Camelids and have been found in deserts, grasslands and savannas throughout paleohistory. Camelids have also Camel developed close relationships with pastoralist and farming cultures wherever they occur. We review the Guanaco evolutionary and paleoecological history of extinct and extant camelids, and then discuss their potential Llama ecological roles within rewilding projects for deserts, grasslands and savannas. The functional ecosystem Rewilding ecology of camelids has not been well researched, and we highlight functions that camelids are likely to Vicuna~ have, but which require further study. -

Giant Camels from the Cenozoic of North America SERIES PUBLICATIONS of the SMITHSONIAN INSTITUTION
Giant Camels from the Cenozoic of North America SERIES PUBLICATIONS OF THE SMITHSONIAN INSTITUTION Emphasis upon publication as a means of "diffusing knowledge" was expressed by the first Secretary of the Smithsonian. In his formal plan for the Institution, Joseph Henry outlined a program that included the following statement: "It is proposed to publish a series of reports, giving an account of the new discoveries in science, and of the changes made from year to year in all branches of knowledge." This theme of basic research has been adhered to through the years by thousands of titles issued in series publications under the Smithsonian imprint, commencing with Smithsonian Contributions to Knowledge in 1848 and continuing with the following active series: Smithsonian Contributions to Anthropology Smithsonian Contributions to Astrophysics Smithsonian Contributions to Botany Smithsonian Contributions to the Earth Sciences Smithsonian Contributions to the H^arine Sciences Smithsonian Contributions to Paleobiology Smithsonian Contributions to Zoology Smithsonian Folklife Studies Smithsonian Studies in Air and Space Smithsonian Studies in History and Technology In these series, the Institution publishes small papers and full-scale monographs that refXJrt the research and collections of its various museums and bureaux or of professional colleagues in the world of science and scholarship. The publications are distributed by mailing lists to libraries, universities, and similar institutions throughout the worid. Papers or monographs submitted for series publication are received by the Smithsonian Institution Press, subject to its own review for format and style, only through departments of the various Smithsonian museums or bureaux, where the manuscripts are given substantive review. Press requirements for manuscript and art preparation are outlined on the inside back cover. -
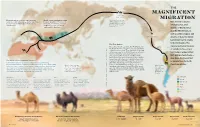
Magnificent Migration
THE MAGNIFICENT Land bridge 8 MY–14,500 Y MIGRATION Domestication of camels began between World camel population today About 6 million years ago, 3,000 and 4,000 years ago—slightly later than is about 30 million: 27 million of camelids began to move Best known today for horses—in both the Arabian Peninsula and these are dromedaries; 3 million westward across the land Camelini western Asia. are Bactrians; and only about that connected Asia and inhabiting hot, arid North America. 1,000 are Wild Bactrians. regions of North Africa and the Middle East, as well as colder steppes and Camelid ancestors deserts of Asia, the family Camelidae had its origins in North America. The The First Camels signature physical features The earliest-known camelids, the Protylopus and Lamini E the Poebrotherium, ranged in sizes comparable to of camels today—one or modern hares to goats. They appeared roughly 40 DMOR I SK million years ago in the North American savannah. two humps, wide padded U L Over the 20 million years that followed, more U L ; feet, well-protected eyes— O than a dozen other ancestral members of the T T O family Camelidae grew, developing larger bodies, . E may have developed K longer legs and long necks to better browse high C I The World's Most Adaptable Traveler? R ; vegetation. Some, like Megacamelus, grew even I as adaptations to North Camels have adapted to some of the Earth’s most demanding taller than the woolly mammoths in their time. LEWSK (Later, in the Middle East, the Syrian camel may American winters. -

Description of a Fossil Camelid from the Pleistocene of Argentina, and a Cladistic Analysis of the Camelinae
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2020 Description of a fossil camelid from the Pleistocene of Argentina, and a cladistic analysis of the Camelinae Lynch, Sinéad ; Sánchez-Villagra, Marcelo R ; Balcarcel, Ana Abstract: We describe a well-preserved South American Lamini partial skeleton (PIMUZ A/V 4165) from the Ensenadan ( 1.95–1.77 to 0.4 Mya) of Argentina. The specimen is comprised of a nearly complete skull and mandible with full tooth rows, multiple elements of anterior and posterior limbs, and a scapula. We tested this specimen’s phylogenetic position and hypothesized it to be more closely related to Lama guanicoe and Vicugna vicugna than to Hemiauchenia paradoxa. We formulate a hypothesis for the placement of PIMUZ A/V 4165 within Camelinae in a cladistic analysis based on craniomandibular and dental characters and propose that future systematic studies consider this specimen as representing a new species. For the first time in a morphological phylogeny, we code terminal taxa at the species levelfor the following genera: Camelops, Aepycamelus, Pleiolama, Procamelus, and Alforjas. Our results indicate a divergence between Lamini and Camelini predating the Barstovian (16 Mya). Camelops appears as monophyletic within the Camelini. Alforjas taylori falls out as a basal member of Camelinae—neither as a Lamini nor Camelini. Pleiolama is polyphyletic, with Pleiolama vera as a basal Lamini and Pleiolama mckennai in a more nested position within the Lamini. Aepycamelus and Procamelus are respectively polyphyletic and paraphyletic. Together, they are part of a group of North American Lamini from the Miocene epoch. -

Information Resources on the South American Camelids: Llamas, Alpacas, Guanacos, and Vicunas 2004-2008
NATIONAL AGRICULTURAL LIBRARY ARCHIVED FILE Archived files are provided for reference purposes only. This file was current when produced, but is no longer maintained and may now be outdated. Content may not appear in full or in its original format. All links external to the document have been deactivated. For additional information, see http://pubs.nal.usda.gov. United States Department of Information Resources on the Agriculture Agricultural Research South American Camelids: Service National Agricultural Llamas, Alpacas, Guanacos, Library Animal Welfare Information Center and Vicunas 2004-2008 AWIC Resource Series No. 12, Revised 2009 AWIC Resource Series No. 12, Revised 2009 United States Information Resources on the Department of Agriculture South American Agricultural Research Service Camelids: Llamas, National Agricultural Alpacas, Guanacos, and Library Animal Welfare Vicunas 2004-2008 Information Center AWIC Resource Series No. 12, Revised 2009 Compiled by: Jean A. Larson, M.S. Animal Welfare Information Center National Agricultural Library U.S. Department of Agriculture Beltsville, Maryland 20705 E-mail: [email protected] Web site: http://awic.nal.usda.gov Available online: http://www.nal.usda.gov/awic/pubs/Camelids/camelids.shtml Disclaimers Te U.S. Department of Agriculture (USDA) prohibits discrimination in all its programs and activities on the basis of race, color, national origin, age, disability, and where applicable, sex, marital status, familial status, parental status, religion, sexual orientation, genetic information, political beliefs, reprisal, or because all or a part of an individual’s income is derived from any public assistance program. (Not all prohibited bases apply to all programs.) Persons with disabilities who require alternative means for communication of program information (Braille, large print, audiotape, etc.) should contact USDA’s TARGET Center at (202) 720-2600 (voice and TDD). -

Spectrum of Camel Evolution and Its Impact on Ancient Human Migration
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 8 February 2021 doi:10.20944/preprints202102.0176.v1 Spectrum of Camel Evolution and its Impact on Ancient Human Migration Indu Sharma1#, Jyotsna Sharma1, 2#, Sachin Kumar1, Hemender Singh2, Varun Sharma1*, Niraj Rai1* 1. Birbal Sahni Institute of Palaeosciences, Lucknow, Uttar Pradesh, 226007, India 2. Shri Mata Vaishno Devi University, Katra, Jammu and Kashmir, 182320, India # First Shared Authors *Corresponding Authors: Dr. Varun Sharma [email protected] Dr. Niraj Rai [email protected] Abstract The Evolutionary history and domestication of Camels are largely unexplored because of the lack of well dated early archaeological records. However, limited records suggest that domestication of Camels likely happened in the late second millennium BCE. Over the time, camels have helped human for their basic needs like meat, milk, wool, dung to long routes transportation. This multifaceted animal has helped the mankind to connect through continents and in trade majorly through the Silk route. In India, both dromedary and Bactrian camels are found and their habitat is entirely different from each other, dromedaries inhabit in hot deserts and Bactrians are found mostly in cold places (Nubra Valley, Ladakh). Fewer studies on Indian dromedaries have been conducted but no such studies are done on Bactrian camels. It is needed to study the genetics of Bactrian camels to find out their genetic affinity and evolutionary history with other Bactrians found in different parts of the world. Furthermore, parallel studies on humans and Bactrian camel are required to understand the co-evolution and migration pattern of humans during their dispersal in different time periods. -

A New Species of Hemiauchenia (Artiodactyla, Camelidae) from the Late Blancan of Florida
Bull. Fla. Mus. Nat. Hist. (2005) 45(4): 435-447 435 A NEW SPECIES OF HEMIAUCHENIA (ARTIODACTYLA, CAMELIDAE) FROM THE LATE BLANCAN OF FLORIDA Julie A. Meachen1 A new species of lamine, Hemiauchenia gracilis n. sp., is present at six late Blancan fossil localities in Florida: Inglis 1A and 1F in Citrus County; DeSoto Shell Pits 1 and 5 in DeSoto County; Santa Fe River 1 in Columbia County; and Waccasassa River 9A in Levy County. The fossil deposits at these locations are thought to be between 1.8 and 2.5 million years old. The holotype specimen is a right mandibular fragment including p4 through m3. Hemiauchenia gracilis lacks p1 through p3. The teeth of H. gracilis are similar to those of Hemiauchenia macrocephala and Hemiauchenia edensis, with an intermediate crown height and a small degree of crenulation. Postcranial elements of H. gracilis are very long and slender, especially the radio-ulna and the phalanges, apparently adaptations for cursoriality. Stable carbon isotope data for H. gracilis show a mixed-feeding signature, with more browse than grass. A phalanx with the size and proportions of those of H. gracilis was found at the late Pliocene 111 Ranch, Graham County, Arizona. This would potentially extend its range as far west as Arizona, drawing it closer toward Mexico and Central America, and possibly giving insight into the origin of the extant Lamini. Key Words: Hemiauchenia gracilis n. sp.; Florida; Pliocene; Lamini; stable carbon isotopes. INTRODUCTION dents and staff. Additional specimens were later found The tribe Lamini (Webb 1965) includes the living llamas, at DeSoto Shell Pits 1 and 5 by Steve and Suzan vicuñas, guanacos, and alpacas. -

Parámetros Genéticos De Caracteres Funcionales Y Secundarios En Alpacas
UNIVERSIDAD COMPLUTENSE DE MADRID FACULTAD DE VETERINARIA Departamento de Producción Animal TESIS DOCTORAL Parámetros genéticos de caracteres funcionales y secundarios en alpacas MEMORIA PARA OPTAR AL GRADO DE DOCTOR PRESENTADA POR Leyfeng Alan Cruz Camacho Director Juan Pablo Gutiérrez García Madrid, 2018 © Leyfeng Alan Cruz Camacho, 2017 Facultad de Veterinaria Departamento de Producción Animal Parámetros Genéticos de Caracteres Funcionales y Secundarios en Alpacas TESIS DOCTORAL Presentada por: Leyfeng Alan Cruz Camacho Director: Dr. Juan Pablo Gutiérrez García Madrid, Febrero del 2017 Facultad de Veterinaria Departamento de Producción Animal PARAMETROS GENÉTICOS DE CARACTERES FUNCIONALES Y SECUNDARIOS EN ALPACAS Memoria presentada por: Leyfeng Alan Cruz Camacho Para optar al grado de Doctor en Veterinaria por la Universidad Complutense de Madrid Dirigida por: Juan Pablo Gutiérrez García Madrid, Febrero del 2017 D. Juan Pablo Gutiérrez García, Profesor Titular del Departamento de Producción Animal de la Facultad de Veterinaria de la Universidad Complutense de Madrid, INFORMA: Que el presente trabajo de investigación titulado “PARÁMETROS GENÉTICOS DE CARACTERES FUNCIONALES Y SECUNDARIOS EN ALPACAS” presentado por, Don Leyfeng Alan Cruz Camacho, y dirigida por quien suscribe, reúne los requisitos necesarios para su exposición y defensa, con el fin de optar al grado de Doctor en Veterinaria. Y para que así conste, firmo el presente informe en Madrid, a 21 de Febrero del 2017 DEDICATORIA A Claudia, por su incondicional apoyo y a mis hijos Adrian y Camila que me alegran cada día, a la memoria de mi abuelita Hermenegilda y mi padre Feliciano. AGRADECIMIENTOS Agradecimientos Agradecer infinitamente a los productores de camélidos del Perú, por el esfuerzo en la reproducción, producción y conservación de tan valioso recurso y por compartirnos sus experiencias, saberes y anécdotas los cuales hemos conceptualizado en esta tesis doctoral. -

Zoonotic Parasites of Dromedary Camels: So Important, So Ignored Alireza Sazmand1* , Anja Joachim2 and Domenico Otranto1,3
Sazmand et al. Parasites Vectors (2019) 12:610 https://doi.org/10.1186/s13071-019-3863-3 Parasites & Vectors REVIEW Open Access Zoonotic parasites of dromedary camels: so important, so ignored Alireza Sazmand1* , Anja Joachim2 and Domenico Otranto1,3 Abstract With a global population of about 35 million in 47 countries, dromedary camels play a crucial role in the economy of many marginal, desert areas of the world where they survive under harsh conditions. Nonetheless, there is scarce knowledge regarding camelsʼ parasite fauna which can reduce their milk and meat productions. In addition, only scattered information is available about zoonotic parasites transmitted to humans via contamination (e.g. Crypto- sporidium spp., Giardia duodenalis, Balantidium coli, Blastocystis spp. and Enterocytozoon bieneusi), as foodborne infections (e.g. Toxoplasma gondii, Trichinella spp. and Linguatula serrata) or by arthropod vectors (Trypanosoma spp.). Herein, we draw attention of the scientifc community and health policy-making organizations to the role camels play in the epidemiology of parasitic zoonotic diseases also in the view of an increase in their farming in desert areas worldwide. Keywords: Camelus dromedarius, Zoonoses, One-Health Background disease transmission to humans, especially in resource- With a worldwide population of about 35 million, cam- poor communities with improper sanitation and medi- els are an important source of meat and milk in many cal access. Tis article reviews the current knowledge on regions of the world, mainly in Africa and Asia [1]. Te zoonotic parasites reported from camels and gaps on the one-humped camel, also known as dromedary (Came- topic that should be addressed in future research. -

Paleontological Contributions
THE UNIVERSITY OF KANSAS PALEONTOLOGICAL CONTRIBUTIONS September 24, 1979 Paper 95 REVISION OF THE CAMELINAE (ARTIODACTYLA, TYLOPODA) AND DESCRIPTION OF THE NEW GENUS ALFORJAS 1 JESSICA A. HARRISON Museum of Natural History, University of Kansas Lawrence, Kansas 66045 ABSTRACT A cladistic analysis of the subfamily Camelinae indicates the presence of two major groups of camels in the Neogene. One group includes the extant lamines of South Amer- ica and their fossil relatives. The other group includes Came/us and its sister group, the gigantic North American genera Megatylo pus and Titanotylo pus. Alforjas taylori, a new genus and species of lamine camel, is described from the Edson local fauna (Hemphillian) of western Kansas. INTRODUCTION The Camelinae constitute one of the most from those of Webb, and are based upon a cla- widely distributed and abundant elements in the distic analysis of the group. Characters used in Neogene faunas of North and South America. In analysis are termed apomorphic (derived) or North America the fossil record of the Camelinae plesiomorphic (primitive), with an autapomor- may be traced from the relatively primitive mid- phic character unique to a particular taxon and a dle Miocene protolabidine camels through the synapomorphic character shared by more than later Cenozoic to the extant genera currently re- one taxon. For more detailed discussions of phy- stricted to Asia and South America. logenetic systematics, see Hennig (1966), Brun- Wortman (1898) produced one of the earliest din (1968), and Wiley (1976); for deductive revisions of the Camelinae, and subsequently sev- testing of hypotheses, see Popper (1968a, b). Mu- eral workers have revised all or part of the group.