Camelus Ferus) Population Using Mitochondrial Markers for Non-Invasively Collected Hair Samples
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Wild Or Bactrian Camel French: German: Wildkamel Spanish: Russian: Dikiy Verblud Chinese
1 of 4 Proposal I / 7 PROPOSAL FOR INCLUSION OF SPECIES ON THE APPENDICES OF THE CONVENTION ON THE CONSERVATION OF MIGRATORY SPECIES OF WILD ANIMALS A. PROPOSAL: Inclusion of the Wild camel Camelus bactrianus in Appendix I of the Convention on the Conservation of Migratory Species of Wild Animals: B. PROPONENT: Mongolia C. SUPPORTING STATEMENT 1. Taxon 1.1. Classis: Mammalia 1.2. Ordo: Tylopoda 1.3. Familia: Camelidae 1.4. Genus: Camelus 1.5. Species: Camelus bactrianus Linnaeus, 1758 1.6. Common names: English: Wild or Bactrian camel French: German: Wildkamel Spanish: Russian: Dikiy verblud Chinese: 2. Biological data 2.1. Distribution Wild populations are restricted to 3 small, remnant populations in China and Mongolia:in the Taklamakan Desert, the deserts around Lop Nur, and the area in and around region A of Mongolia’s Great Gobi Strict Protected Area (Reading et al 2000). In addition, there is a small semi-captive herd of wild camels being maintained and bred outside of the Park. 2.2. Population Surveys over the past several decades have suggested a marked decline in wild bactrian camel numbers and reproductive success rates (Zhirnov and Ilyinsky 1986, Anonymous 1988, Tolgat and Schaller 1992, Tolgat 1995). Researchers suggest that fewer than 500 camels remain in Mongolia and that their population appears to be declining (Xiaoming and Schaller 1996). Globally, scientists have recently suggested that less than 900 individuals survive in small portions of Mongolia and China (Tolgat and Schaller 1992, Hare 1997, Tolgat 1995, Xiaoming and Schaller 1996). However, most of the population estimates from both China and Mongolia were made using methods which preclude rigorous population estimation. -

Camelids: New Players in the International Animal Production Context
Tropical Animal Health and Production (2020) 52:903–913 https://doi.org/10.1007/s11250-019-02197-2 REVIEWS Camelids: new players in the international animal production context Mousa Zarrin1 & José L. Riveros2 & Amir Ahmadpour1,3 & André M. de Almeida4 & Gaukhar Konuspayeva5 & Einar Vargas- Bello-Pérez6 & Bernard Faye7 & Lorenzo E. Hernández-Castellano8 Received: 30 October 2019 /Accepted: 22 December 2019 /Published online: 2 January 2020 # Springer Nature B.V. 2020 Abstract The Camelidae family comprises the Bactrian camel (Camelus bactrianus), the dromedary camel (Camelus dromedarius), and four species of South American camelids: llama (Lama glama),alpaca(Lama pacos)guanaco(Lama guanicoe), and vicuña (Vicugna vicugna). The main characteristic of these species is their ability to cope with either hard climatic conditions like those found in arid regions (Bactrian and dromedary camels) or high-altitude landscapes like those found in South America (South American camelids). Because of such interesting physiological and adaptive traits, the interest for these animals as livestock species has increased considerably over the last years. In general, the main animal products obtained from these animals are meat, milk, and hair fiber, although they are also used for races and work among other activities. In the near future, climate change will likely decrease agricultural areas for animal production worldwide, particularly in the tropics and subtropics where competition with crops for human consumption is a major problem already. In such conditions, extensive animal production could be limited in some extent to semi-arid rangelands, subjected to periodical draughts and erratic patterns of rainfall, severely affecting conventional livestock production, namely cattle and sheep. -

Bactrian Camel, Two-Humped Camel
Camelus ferus/bactrianus Common name: Bactrian camel, two-humped camel Local name: Havtagai (Mongolian), Wildkamel (German), Jya nishpa yapung (Ladakhi) Classification: Kingdom: Animalia Phylum: Chordata Class: Mammalia Order: Artiodactyla Family: Camelidae Genus: Camelus Species: ferus/bactrianus Profile: The scientific name of the wild Bactrian camel is Camelus ferus, while the domesticated form is called Camelus bactrianus. The distinctive feature of the animal is that it is two-humped whereas the Dromedary camel has a single hump. DNA tests have revealed that there are two or three distinct genetic differences and about 3% base difference between the wild and domestic populations of Bactrian camels. They also differ physically. The wild Bactrian camel is smaller and slender than the domestic breed. The wild camels have a sandy gray- brown coat while the domestic ones have a dark brown coat. The predominant difference between them however is the shape of the humps. While that of the wild camel are small and pyramid-like, those of the domestic ones are large and irregular. The face of a Bactrian camel is long and triangular with a split upper lip. The Bactrian camel is highly adapted to surviving the cold desert climate. Each foot has an undivided sole with two large toes that can spread wide apart for walking on sand. The ears and nose are lined with hair to protect against sand and the muscular nostrils can be closed during sandstorms. The eyes are protected from sand and debris by a double layer of long eyelashes while bushy eyebrows give protection from the sun. It grows a thick shaggy coat during winter, which is shed very rapidly in spring to give the animal a shorn look. -

Characterization of Caseins from Mongolian Yak, Khainak, and Bactrian Camel B Ochirkhuyag, Jm Chobert, M Dalgalarrondo, Y Choiset, T Haertlé
Characterization of caseins from Mongolian yak, khainak, and bactrian camel B Ochirkhuyag, Jm Chobert, M Dalgalarrondo, Y Choiset, T Haertlé To cite this version: B Ochirkhuyag, Jm Chobert, M Dalgalarrondo, Y Choiset, T Haertlé. Characterization of caseins from Mongolian yak, khainak, and bactrian camel. Le Lait, INRA Editions, 1997, 77 (5), pp.601-613. hal-00929550 HAL Id: hal-00929550 https://hal.archives-ouvertes.fr/hal-00929550 Submitted on 1 Jan 1997 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Lait (1997) 77, 601-613 601 © Eisevier/Inra Original article Characterization of caseins from Mongolian yak, khainak, and bactrian cam el B Ochirkhuyag 2, lM Chobert 1*, M Dalgalarrondo 1, Y Choiset 1, T Haertlé ' 1 Laboratoire d'étude des interactions des molécules alimentaires, Inra, rue de la Géraudière, BP 71627, 44316 Nantes cedex 03, France; 2 Institute of Chemistry, Academy of Sciences, Vlan Bator, Mongolia (Received 25 November 1996; accepted 5 May 1997) Summary - The composition of acid-precipitated caseins from ruminant Mongolian domestic ani- maIs was analyzed and a comparative study between camel (Camelus bactrianus) and dromedary (Camelus dromedarius) was realized. Acid-precipitated whole caseins were analyzed for ami no acid composition, separated by anion exchange chromatography and identified by alkaline urea-PAGE. -

Bison Literature Review Biology
Bison Literature Review Ben Baldwin and Kody Menghini The purpose of this document is to compare the biology, ecology and basic behavior of cattle and bison for a management context. The literature related to bison is extensive and broad in scope covering the full continuum of domestication. The information incorporated in this review is focused on bison in more or less “wild” or free-ranging situations i.e.., not bison in close confinement or commercial production. While the scientific literature provides a solid basis for much of the basic biology and ecology, there is a wealth of information related to management implications and guidelines that is not captured. Much of the current information related to bison management, behavior (especially social organization) and practical knowledge is available through local experts, current research that has yet to be published, or popular literature. These sources, while harder to find and usually more localized in scope, provide crucial information pertaining to bison management. Biology Diet Composition Bison evolutional history provides the basis for many of the differences between bison and cattle. Bison due to their evolution in North America ecosystems are better adapted than introduced cattle, especially in grass dominated systems such as prairies. Many of these areas historically had relatively low quality forage. Bison are capable of more efficient digestion of low-quality forage then cattle (Peden et al. 1973; Plumb and Dodd 1993). Peden et al. (1973) also found that bison could consume greater quantities of low protein and poor quality forage then cattle. Bison and cattle have significant dietary overlap, but there are slight differences as well. -

Convention on Migratory Species
CMS Distribution: General CONVENTION ON UNEP/CMS/COP11/Inf.21 MIGRATORY 16 July 2014 SPECIES Original: English 11th MEETING OF THE CONFERENCE OF THE PARTIES Quito, Ecuador, 4-9 November 2014 Agenda Item 23.3.1 ASSESSMENT OF GAPS AND NEEDS IN MIGRATORY MAMMALS CONSERVATION IN CENTRAL ASIA 1. In response to multiple mandates (notably Concerted and Cooperative Actions, Rec.8.23 and 9.1, Res.10.3 and 10.9), CMS has strengthened its work for the conservation of large mammals in the central Asian region and inter alia initiated a gap analysis and needs assessment, including status reports of prioritized central Asian migratory mammals to obtain a better picture of the situation in the region and to identify priorities for conservation. Range States and a large number of relevant experts were engaged in the process, and national stakeholder consultation meetings organized in several countries. 2. The Meeting Document along with the Executive Summary of the assessment is available as UNEP/CMS/COP11/Doc.23.3.1. For reasons of economy, documents are printed in a limited number, and will not be distributed at the Meeting. Delegates are requested to bring their copy to the meeting and not to request additional copies. UNEP/CMS/COP11/Inf.21 Assessment of gaps and needs in migratory mammal conservation in Central Asia Report prepared for the Convention on the Conservation of Migratory Species of Wild Animals (CMS) and the Deutsche Gesellschaft für Internationale Zusammenarbeit (GIZ) GmbH. Financed by the Ecosystem Restoration in Central Asia (ERCA) component of the European Union Forest and Biodiversity Governance Including Environmental Monitoring Project (FLERMONECA). -

Last Interglacial (MIS 5) Ungulate Assemblage from the Central Iberian Peninsula: the Camino Cave (Pinilla Del Valle, Madrid, Spain)
Palaeogeography, Palaeoclimatology, Palaeoecology 374 (2013) 327–337 Contents lists available at SciVerse ScienceDirect Palaeogeography, Palaeoclimatology, Palaeoecology journal homepage: www.elsevier.com/locate/palaeo Last Interglacial (MIS 5) ungulate assemblage from the Central Iberian Peninsula: The Camino Cave (Pinilla del Valle, Madrid, Spain) Diego J. Álvarez-Lao a,⁎, Juan L. Arsuaga b,c, Enrique Baquedano d, Alfredo Pérez-González e a Área de Paleontología, Departamento de Geología, Universidad de Oviedo, C/Jesús Arias de Velasco, s/n, 33005 Oviedo, Spain b Centro Mixto UCM-ISCIII de Evolución y Comportamiento Humanos, C/Sinesio Delgado, 4, 28029 Madrid, Spain c Departamento de Paleontología, Facultad de Ciencias Geológicas, Universidad Complutense de Madrid, Ciudad Universitaria, 28040 Madrid, Spain d Museo Arqueológico Regional de la Comunidad de Madrid, Plaza de las Bernardas, s/n, 28801-Alcalá de Henares, Madrid, Spain e Centro Nacional de Investigación sobre la Evolución Humana (CENIEH), Paseo Sierra de Atapuerca, s/n, 09002 Burgos, Spain article info abstract Article history: The fossil assemblage from the Camino Cave, corresponding to the late MIS 5, constitutes a key record to un- Received 2 November 2012 derstand the faunal composition of Central Iberia during the last Interglacial. Moreover, the largest Iberian Received in revised form 21 January 2013 fallow deer fossil population was recovered here. Other ungulate species present at this assemblage include Accepted 31 January 2013 red deer, roe deer, aurochs, chamois, wild boar, horse and steppe rhinoceros; carnivores and Neanderthals Available online 13 February 2013 are also present. The origin of the accumulation has been interpreted as a hyena den. Abundant fallow deer skeletal elements allowed to statistically compare the Camino Cave fossils with other Keywords: Early Late Pleistocene Pleistocene and Holocene European populations. -

1 BOARD of ANIMAL HEALTH Subpart 2 Chapter 12 Sheep And
BOARD OF ANIMAL HEALTH Subpart 2 Chapter 12 Sheep and Goats 109 All sheep and goats, except those for immediate slaughter shall be accompanied by an official certificate of veterinary inspection (OCVI) and shall comply with the following: 1. Intact sheep and goats require individual identification by an official USDA Scrapie eartag, brand, or tattoo recorded on the OCVI. 2. “I certify these animals are free of clinical signs of the diseases contagious footrot, keratoconjunctivitis, contagious ecthyma (Orf), scabies and lice and that the sexually intact animals represented on this form are not known to be scrapie- positive, suspect, high risk, or exposed, and did not originate from a known infected, source, exposed, or noncompliant flock.” 3. When originating from an area known to have scabies, must be dipped within ten (10) days immediately preceding the date of entry in an USDA approved dip, and maintained on absolutely clean premises until delivered to the final destination. Dairy goats and dairy sheep maintained separate from other sheep and goats are exempt from dipping when certified free of scabies on OCVI. 4. Dairy goats and dairy sheep over 6 months of age must be negative to an official tuberculin test and an official brucellosis test made within 30 days immediately preceding date of entry. 5. All sheep and goats for immediate slaughter shall be consigned to a recognized slaughtering establishment on either an OCVI or permit or waybill or inspection certification from federally inspected stockyards. In either instance, a copy shall accompany sheep and goats and a copy shall be forwarded to the State Veterinarian of Mississippi. -

Pleistocene Mammals from Extinction Cave, Belize
Canadian Journal of Earth Sciences Pleistocene Mammals From Extinction Cave, Belize Journal: Canadian Journal of Earth Sciences Manuscript ID cjes-2018-0178.R3 Manuscript Type: Article Date Submitted by the 04-May-2019 Author: Complete List of Authors: Churcher, C.S.; University of Toronto, Zoology Central America, Pleistocene, Fauna, Vertebrate Palaeontology, Keyword: Limestone cave Is the invited manuscript for consideration in a Special Not applicableDraft (regular submission) Issue? : https://mc06.manuscriptcentral.com/cjes-pubs Page 1 of 43 Canadian Journal of Earth Sciences 1 1 PLEISTOCENE MAMMALS FROM EXTINCTION CAVE, BELIZE 2 by C.S. CHURCHER1 Draft 1Department of Zoology, University of Toronto, Toronto, Ontario Canada M5S 2C6 and 322-240 Dallas Rd., Victoria, British Columbia, Canada V8V 4X9 (corresponding address): e-mail [email protected] https://mc06.manuscriptcentral.com/cjes-pubs Canadian Journal of Earth Sciences Page 2 of 43 2 4 5 ABSTRACT. A small mammalian fauna is recorded from Extinction Cave (also called Sibun 6 Cave), east of Belmopan, on the Sibun River, Belize, Central America. The animals recognized 7 are armadillo (†Dasypus bellus), American lion (†Panthera atrox), jaguar (P. onca), puma or 8 mountain lion (Puma concolor), Florida spectacled bear (†Tremarctos floridanus), javelina or 9 collared peccary (Pecari tajacu), llama (Camelidae indet., ?†Palaeolama mirifica), red brocket 10 deer (Mazama americana), bison (Bison sp.) and Mexican half-ass (†Equus conversidens), and 11 sabre-tooth cat († Smilodon fatalis) may also be represented (‘†’ indicates an extinct taxon). 12 Bear and bison are absent from the region today. The bison record is one of the more southernly 13 known. The bear record is almost the mostDraft westerly known and a first for Central America. -
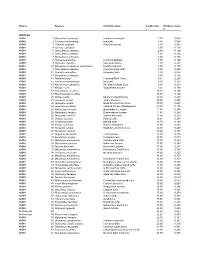
PDF File Containing Table of Lengths and Thicknesses of Turtle Shells And
Source Species Common name length (cm) thickness (cm) L t TURTLES AMNH 1 Sternotherus odoratus common musk turtle 2.30 0.089 AMNH 2 Clemmys muhlenbergi bug turtle 3.80 0.069 AMNH 3 Chersina angulata Angulate tortoise 3.90 0.050 AMNH 4 Testudo carbonera 6.97 0.130 AMNH 5 Sternotherus oderatus 6.99 0.160 AMNH 6 Sternotherus oderatus 7.00 0.165 AMNH 7 Sternotherus oderatus 7.00 0.165 AMNH 8 Homopus areolatus Common padloper 7.95 0.100 AMNH 9 Homopus signatus Speckled tortoise 7.98 0.231 AMNH 10 Kinosternon subrabum steinochneri Florida mud turtle 8.90 0.178 AMNH 11 Sternotherus oderatus Common musk turtle 8.98 0.290 AMNH 12 Chelydra serpentina Snapping turtle 8.98 0.076 AMNH 13 Sternotherus oderatus 9.00 0.168 AMNH 14 Hardella thurgi Crowned River Turtle 9.04 0.263 AMNH 15 Clemmys muhlenbergii Bog turtle 9.09 0.231 AMNH 16 Kinosternon subrubrum The Eastern Mud Turtle 9.10 0.253 AMNH 17 Kinixys crosa hinged-back tortoise 9.34 0.160 AMNH 18 Peamobates oculifers 10.17 0.140 AMNH 19 Peammobates oculifera 10.27 0.140 AMNH 20 Kinixys spekii Speke's hinged tortoise 10.30 0.201 AMNH 21 Terrapene ornata ornate box turtle 10.30 0.406 AMNH 22 Terrapene ornata North American box turtle 10.76 0.257 AMNH 23 Geochelone radiata radiated tortoise (Madagascar) 10.80 0.155 AMNH 24 Malaclemys terrapin diamondback terrapin 11.40 0.295 AMNH 25 Malaclemys terrapin Diamondback terrapin 11.58 0.264 AMNH 26 Terrapene carolina eastern box turtle 11.80 0.259 AMNH 27 Chrysemys picta Painted turtle 12.21 0.267 AMNH 28 Chrysemys picta painted turtle 12.70 0.168 AMNH 29 -

Redalyc.Bienestar Animal En Bovinos De Leche: Selección De Indicadores
RIA. Revista de Investigaciones Agropecuarias ISSN: 0325-8718 [email protected] Instituto Nacional de Tecnología Agropecuaria Argentina MARTÍNEZ, G. M.; SUÁREZ, V. H.; GHEZZI, M. D. Bienestar animal en bovinos de leche: selección de indicadores vinculados a la salud y producción RIA. Revista de Investigaciones Agropecuarias, vol. 42, núm. 2, agosto, 2016, pp. 153- 160 Instituto Nacional de Tecnología Agropecuaria Buenos Aires, Argentina Disponible en: http://www.redalyc.org/articulo.oa?id=86447075008 Cómo citar el artículo Número completo Sistema de Información Científica Más información del artículo Red de Revistas Científicas de América Latina, el Caribe, España y Portugal Página de la revista en redalyc.org Proyecto académico sin fines de lucro, desarrollado bajo la iniciativa de acceso abierto Agosto 2016, Argentina 15 Bienestar animal en bovinos de leche: selección de indicadores vinculados a la salud y producción MARTÍNEZ, G. M. 1*; SUÁREZ, V. H. 2; GHEZZI, M. D. 3 RESUMEN El bienestar animal ha sido denido por la Organización Mundial de Sanidad Animal (OIE) como el término amplio que describe la manera en que los individuos se enfrentan con el ambiente y que incluye su sanidad, sus percepciones, su estado anímico y otros e fectos positivos o negativos que inuyen sobre los mecanis - mos físicos y psíquicos del animal. Durante años se tuvo como objetivo principal dentro de los programas de mejora genética en rodeos lecheros el aumento en la producción de leche por individuo; posteriormente se trabajó en compatibilizar ese incremento en el rinde con una mayor eciencia en la conversión alimenticia. A lo largo de este período todo el sistema productivo se fue transformando de manera tal de ofrecerle a esos animales de alto mérito genético el ambiente necesario para que consiguiesen expresar su potencial. -

{TEXTBOOK} Is a Camel a Mammal?
IS A CAMEL A MAMMAL? PDF, EPUB, EBOOK Tish Rabe,Jim Durk | 48 pages | 04 Jun 2001 | HarperCollins Publishers | 9780007111077 | English | London, United Kingdom Is a Camel a Mammal? PDF Book Ano ang katangian ng salawikain? Retrieved 5 December Camel is an animal and is not an egg laying mammal. So we had what amounted to two pounds or more of rubber for dinner that night. Is camel a marsupial mammal? What rhymes with mammal? Center for Muslim-Jewish Engagement. Collared peccary P. The Oxford Companion to Food 2nd ed. Both the dromedary the seven-humped camel of Arabia and the Bactrian camel the two-humped camel of Central Asia had been domesticated since before BC. Red brocket M. In addition to providing the Roman Army with its best archers, the Easterners largely Arabs but generally known as 'Syrians' served as Rome's most effective dromedarii or camel-mounted troops. Even salty water can be tolerated, and between drinks it forages far from oases to find food unavailable to other livestock. Somalia a Country Study. White-tailed deer O. Namespaces Article Talk. Do camels lay eggs? Greenwood Publishing Group. View 1 comment. The reason why Cyrus opposed his camels to the enemy's horse was because the horse has a natural dread of the camel, and cannot abide either the sight or the smell of that animal. Archived from the original on 4 August Melissa Stewart. Camel Corps experiment. Is the word camel a short vowel word? ABC News. Consequently, these schools hold that Muslims must perform wudhu ablution before the next time they pray after eating camel meat.