Magnetic Resonance Imaging Findings in Adult-Form Myotonic Dystrophy Type 1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CADASIL Testing
Lab Management Guidelines V1.0.2020 CADASIL Testing MOL.TS.144.A v1.0.2020 Introduction CADASIL testing is addressed by this guideline. Procedures addressed The inclusion of any procedure code in this table does not imply that the code is under management or requires prior authorization. Refer to the specific Health Plan's procedure code list for management requirements. Procedures addressed by this Procedure codes guideline NOTCH3 Known Familial Mutation 81403 Analysis NOTCH3 Targeted Sequencing 81406 NOTCH3 Deletion/Duplication Analysis 81479 What is CADASIL Definition CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy) is an adult-onset form of cerebrovascular disease. There are no generally accepted clinical diagnostic criteria for CADASIL and symptoms vary among affected individuals. Signs and symptoms Typical signs and symptoms include1,2,3 Transient ischemic attacks and ischemic stroke, occurs at a mean age of 47 years (age range 20-70 years), in most cases without conventional vascular risk factors cognitive disturbance, primarily affecting executive function, may start as early as age 35 years psychiatric or behavioral abnormalities migraine with aura, occurs with a mean age of onset of 30 years (age range 6-48 years), and Less common symptoms include: © 2020 eviCore healthcare. All Rights Reserved. 1 of 7 400 Buckwalter Place Boulevard, Bluffton, SC 29910 (800) 918-8924 www.eviCore.com Lab Management Guidelines V1.0.2020 recurrent seizures with onset in middle age, usually secondary to stroke acute encephalopathy, with a mean age of onset of 42 years Life expectancy for men with CADASIL is reduced by approximately five years and for women by 1 to 2 years.4 Diagnosis Brain Magnetic Resonance Imaging (MRI) findings include T2-signal-abnormalities in the white matter of the temporal pole and T2-signal-abnormalities in the external capsule and corpus callosum.1,2 CADASIL is suspected in an individual with the clinical signs and MRI findings. -

The National Economic Burden of Rare Disease Study February 2021
Acknowledgements This study was sponsored by the EveryLife Foundation for Rare Diseases and made possible through the collaborative efforts of the national rare disease community and key stakeholders. The EveryLife Foundation thanks all those who shared their expertise and insights to provide invaluable input to the study including: the Lewin Group, the EveryLife Community Congress membership, the Technical Advisory Group for this study, leadership from the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH), the Undiagnosed Diseases Network (UDN), the Little Hercules Foundation, the Rare Disease Legislative Advocates (RDLA) Advisory Committee, SmithSolve, and our study funders. Most especially, we thank the members of our rare disease patient and caregiver community who participated in this effort and have helped to transform their lived experience into quantifiable data. LEWIN GROUP PROJECT STAFF Grace Yang, MPA, MA, Vice President Inna Cintina, PhD, Senior Consultant Matt Zhou, BS, Research Consultant Daniel Emont, MPH, Research Consultant Janice Lin, BS, Consultant Samuel Kallman, BA, BS, Research Consultant EVERYLIFE FOUNDATION PROJECT STAFF Annie Kennedy, BS, Chief of Policy and Advocacy Julia Jenkins, BA, Executive Director Jamie Sullivan, MPH, Director of Policy TECHNICAL ADVISORY GROUP Annie Kennedy, BS, Chief of Policy & Advocacy, EveryLife Foundation for Rare Diseases Anne Pariser, MD, Director, Office of Rare Diseases Research, National Center for Advancing Translational Sciences (NCATS), National Institutes of Health Elisabeth M. Oehrlein, PhD, MS, Senior Director, Research and Programs, National Health Council Christina Hartman, Senior Director of Advocacy, The Assistance Fund Kathleen Stratton, National Academies of Science, Engineering and Medicine (NASEM) Steve Silvestri, Director, Government Affairs, Neurocrine Biosciences Inc. -

Cadasil Pathogenesis, Clinical and Radiological Findings and Treatment
View and review Arq Neuropsiquiatr 2010;68(2):287-299 Cadasil Pathogenesis, clinical and radiological findings and treatment Charles André ABSTRACT Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is the most common genetic cause of ischemic strokes and a most important model for the study of subcortical vascular dementia. This unrelentlessly progressive disease affects many hundreds of families all over the world but is not well studied in Brazil. This manuscript reviews pathogenetic, clinical, radiological and therapeutic features of CADASIL. The causal mutations are now very well known, but the same can not be said about its intimate pathogenetic mechanisms. The variable clinical presentation should lead physicians to actively pursue the diagnosis in many settings and to more thouroughly investigate family history in first degree relatives. A rational approach to genetic testing is however needed. Treatment of CADASIL is still largely empiric. High- quality therapeutic studies involving medications and cognitive interventions are strongly needed in CADASIL. Key words: CADASIL, etiology, genetics, diagnosis, therapeutics. CADASIL: patogênese, achados clínicos e radiológicos e tratamento RESUMO CADASIL é a causa genética mais freqüente de infartos cerebrais e constitui modelo importante de estudo de demências vasculares subcorticais. De natureza inexoravelmente progressiva, afeta milhares de pessoas em todo o mundo. Sua importância é pouco reconhecida entre nós, o que nos levou à presente revisão dos principais aspectos patogenéticos, clínicos, neuroradiológicos e terapêuticos da doença. As mutações causais são hoje bem conhecidas, mas os mecanismos patogenéticos íntimos ainda permanecem misteriosos. A apresentação clínica variável deve fazer com que os médicos considerem o diagnóstico em vários contextos clínicos e investiguem de forma mais extensa que o usual a história familial deparentes de primeiro grau. -

Prevalence and Incidence of Rare Diseases: Bibliographic Data
Number 1 | January 2019 Prevalence and incidence of rare diseases: Bibliographic data Prevalence, incidence or number of published cases listed by diseases (in alphabetical order) www.orpha.net www.orphadata.org If a range of national data is available, the average is Methodology calculated to estimate the worldwide or European prevalence or incidence. When a range of data sources is available, the most Orphanet carries out a systematic survey of literature in recent data source that meets a certain number of quality order to estimate the prevalence and incidence of rare criteria is favoured (registries, meta-analyses, diseases. This study aims to collect new data regarding population-based studies, large cohorts studies). point prevalence, birth prevalence and incidence, and to update already published data according to new For congenital diseases, the prevalence is estimated, so scientific studies or other available data. that: Prevalence = birth prevalence x (patient life This data is presented in the following reports published expectancy/general population life expectancy). biannually: When only incidence data is documented, the prevalence is estimated when possible, so that : • Prevalence, incidence or number of published cases listed by diseases (in alphabetical order); Prevalence = incidence x disease mean duration. • Diseases listed by decreasing prevalence, incidence When neither prevalence nor incidence data is available, or number of published cases; which is the case for very rare diseases, the number of cases or families documented in the medical literature is Data collection provided. A number of different sources are used : Limitations of the study • Registries (RARECARE, EUROCAT, etc) ; The prevalence and incidence data presented in this report are only estimations and cannot be considered to • National/international health institutes and agencies be absolutely correct. -

The Myotonic Dystrophies: Diagnosis and Management Chris Turner,1 David Hilton-Jones2
Review J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.2008.158261 on 22 February 2010. Downloaded from The myotonic dystrophies: diagnosis and management Chris Turner,1 David Hilton-Jones2 1Department of Neurology, ABSTRACT asymptomatic relatives as well as prenatal and National Hospital for Neurology There are currently two clinically and molecularly defined preimplantation diagnosis can also be performed.7 and Neurosurgery, London, UK 2Department of Clinical forms of myotonic dystrophy: (1) myotonic dystrophy Neurology, The Radcliffe type 1 (DM1), also known as ‘Steinert’s disease’; and Anticipation Infirmary, Oxford, UK (2) myotonic dystrophy type 2 (DM2), also known as DMPK alleles greater than 37 CTG repeats in length proximal myotonic myopathy. DM1 and DM2 are are unstable and may expand in length during meiosis Correspondence to progressive multisystem genetic disorders with several and mitosis. Children of a parent with DM1 may Dr C Turner, Department of Neurology, National Hospital for clinical and genetic features in common. DM1 is the most inherit repeat lengths considerably longer than those Neurology and Neurosurgery, common form of adult onset muscular dystrophy whereas present in the transmitting parent. This phenomenon Queen Square, London WC1N DM2 tends to have a milder phenotype with later onset of causes ‘anticipation’, which is the occurrence of 3BG, UK; symptoms and is rarer than DM1. This review will focus increasing disease severity and decreasing age of onset [email protected] on the clinical features, diagnosis and management of in successive generations. The presence of a larger Received 1 December 2008 DM1 and DM2 and will briefly discuss the recent repeat leads to earlier onset and more severe disease Accepted 18 December 2008 advances in the understanding of the molecular and causes the more severe phenotype of ‘congenital’ pathogenesis of these diseases with particular reference DM1 (figure 2).8 9 A child with congenital DM 1 to new treatments using gene therapy. -

Insurance and Advance Pay Test Requisition
Insurance and Advance Pay Test Requisition (2021) For Specimen Collection Service, Please Fax this Test Requisition to 1.610.271.6085 Client Services is available Monday through Friday from 8:30 AM to 9:00 PM EST at 1.800.394.4493, option 2 Patient Information Patient Name Patient ID# (if available) Date of Birth Sex designated at birth: 9 Male 9 Female Street address City, State, Zip Mobile phone #1 Other Phone #2 Patient email Language spoken if other than English Test and Specimen Information Consult test list for test code and name Test Code: Test Name: Test Code: Test Name: 9 Check if more than 2 tests are ordered. Additional tests should be checked off within the test list ICD-10 Codes (required for billing insurance): Clinical diagnosis: Age at Initial Presentation: Ancestral Background (check all that apply): 9 African 9 Asian: East 9 Asian: Southeast 9 Central/South American 9 Hispanic 9 Native American 9 Ashkenazi Jewish 9 Asian: Indian 9 Caribbean 9 European 9 Middle Eastern 9 Pacific Islander Other: Indications for genetic testing (please check one): 9 Diagnostic (symptomatic) 9 Predictive (asymptomatic) 9 Prenatal* 9 Carrier 9 Family testing/single site Relationship to Proband: If performed at Athena, provide relative’s accession # . If performed at another lab, a copy of the relative’s report is required. Please attach detailed medical records and family history information Specimen Type: Date sample obtained: __________ /__________ /__________ 9 Whole Blood 9 Serum 9 CSF 9 Muscle 9 CVS: Cultured 9 Amniotic Fluid: Cultured 9 Saliva (Not available for all tests) 9 DNA** - tissue source: Concentration ug/ml Was DNA extracted at a CLIA-certified laboratory or a laboratory meeting equivalent requirements (as determined by CAP and/or CMS)? 9 Yes 9 No 9 Other*: If not collected same day as shipped, how was sample stored? 9 Room temp 9 Refrigerated 9 Frozen (-20) 9 Frozen (-80) History of blood transfusion? 9 Yes 9 No Most recent transfusion: __________ /__________ /__________ *Please contact us at 1.800.394.4493, option 2 prior to sending specimens. -

X-Linked Myotubular Myopathy and Chylothorax
ARTICLE IN PRESS Neuromuscular Disorders xxx (2007) xxx–xxx www.elsevier.com/locate/nmd Case report X-linked myotubular myopathy and chylothorax Koenraad Smets * Department of Neonatology, Ghent University Hospital, De Pintelaan 185, B-9000 Ghent, Belgium Received 10 August 2007; received in revised form 4 October 2007; accepted 24 October 2007 Abstract X-linked myotubular myopathy usually presents at birth with hypotonia and respiratory distress. Phenotypic presentation, however, can be extreme variable. We report on a newborn baby, who presented with the severe form of the disease. In the second week of life, he developed a clinically relevant chylothorax, needing drainage and treatment with octreotide acetate. Pleural effusions are frequently described in patients with congenital myotonic dystrophy. To our knowledge, the association of chylothorax and X-linked myotubular myopathy has not been described to date. As chylothorax could not be attributed to any evident condition in this child, perhaps it may be added to the clinical spectrum of X-linked myotubular myopathy. Ó 2007 Elsevier B.V. All rights reserved. Keywords: X-linked myotubular myopathy; Chylothorax 1. Introduction drainage (Fig. 1). Laboratory examination was compatible with chylothorax (5230 white blood cells/ll, 98% lympho- Congenital myopathies often present with hypotonia cytes; chylomicrons were present; triglycerides 746 mg/dl). and respiratory distress from birth, although their expres- There were no central venous catheters in place who could sion may be delayed. In most cases muscle biopsy is war- have caused thrombosis, impairing lymphatic flow, neither ranted for definitive diagnosis. In some instances could any other risk factor for chylothorax be identified. -

Genetics of Hypertension Paul N
November/December 2003 ⅐ Vol. 5 ⅐ No. 6 review Genetics of hypertension Paul N. Hopkins, MD, MSPH and Steven C. Hunt, PhD Hypertension is the most prevalent cardiovascular disorder. progressively to arterial and arteriolar hypertrophy, arterio- In the 1999 to 2000 NHANES survey, the prevalence of hyper- sclerosis and arteriolosclerosis, and with very high pressures to tension progressively increased from 7.2% in those aged 18 to fibrinoid change and fibrinoid necrosis in arterioles. These lat- 39 to 30.1% in 40 to 59 year olds and 65.4% in those 60 and ter changes can result in lumen compromise of arterioles re- older.1 Risk of both coronary atherosclerosis and stroke in- sulting in lacunar stroke, Charcot-Bouchard aneurysms, glo- crease exponentially as blood pressure rises (see Fig. 1).2 Al- merulosclerosis and nephrosclerosis, and ultimately malignant though the relative risk for stroke increases more rapidly than hypertension in the kidney and retinal ischemia and blindness. coronary disease, at any pressure, the absolute risk for coro- Risk of intracerebral hemorrhage is increased 33-fold at stage 3 nary disease is considerably greater than for stroke. An insight or higher pressures compared to normal blood pressure.23 Un- into this finding comes from autopsy studies that show that the treated, malignant hypertension is associated with a 5-year carotid and intracerebral vascular beds are relatively protected mortality rate of 95% with 65% dying from congestive heart from atherosclerosis as compared to the coronary circulation, failure, -
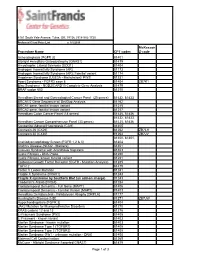
Procedure Name CPT Codes Mckesson Z-Code Achondroplasia
6161 South Yale Avenue, Tulsa, OK, 74136 | 918-502-1720 Preferred Client Price List v. 1/1/2019 McKesson Procedure Name CPT codes Z-code Achondroplasia {FGFR 3} 81401 Albright Hereditary Osteodystrophy {GNAS1} 81479 Amyotrophic Lateral Sclerosis {SOD1} 81404 Androgen Insensitivity Syndrome {AR} 81173 Androgen Insensitivity Syndrome {AR}; Familial variant 81174 Angelman Syndrome {UBE3A - Methylation}/ PWS 81331 Apert Syndrome - FGFR2 exon 8 81404 ZB7K1 Blau Syndrome - NOD2/CARD15 Complete Gene Analysis 81479 BRAF codon 600 81210 Hereditary Breast and Gynecological Cancer Panel (25 genes) 81432, 81433 BRCA1/2 Gene Sequence w/ Del/Dup Analysis 81162 BRCA1 gene, familial known variant 81215 BRCA2 gene, familial known variant 81217 Hereditary Colon Cancer Panel (18 genes) 81435, 81436 81432, 81433, Hereditary Cancer Comprehensive Panel (33 genes) 81435, 81436 Congenital Adrenal Hyperplasia {CAH} 81405 Connexin 26 {CX26} 81252 ZB7LH Connexin 30 {CX30} 81254 ZB7JV 81400, 81401, Craniodysmorphology Screen {FGFR 1,2 & 3} 81404 Crohn's Disease {NOD2 - Markers} 81401 Crouzon Syndrome with Acanthosis Nigricans 81403 Cystic Fibrosis - DNA Probe 81220 Cystic Fibrosis, known familial variant 81221 Epidermal Growth Factor Receptor {EGFR - Mutation Analysis} 81235 FGFR 2 81479 Factor V Leiden Mutation 81241 Fragile X Syndrome {FRAX1} 81243 Fragile X syndrome by Southern Blot (an add-on charge) 81243 Friederich's Ataxia {FRDA} 81284 Frontotemporal Dementia - Full Gene (MAPT) 81406 Frontotemporal Dementia - Familial Variant (MAPT) 81403 Hereditary Dentatorubral -

New Insights Into the Diagnosis and Treatment of Single-Gene Disorders Associated with Cryptogenic Ischemic Stroke
New Insights Into the Diagnosis and Treatment of Single-Gene Disorders Associated With Cryptogenic Ischemic Stroke A Continuing Medical Education Monograph Release date: September 30, 2010 Expiration date: September 30, 2013 Editor Contributors Katherine B. Sims, MD Mark J. Alberts, MD, FAHA Louis R. Caplan, MD Massachusetts General Hospital Northwestern University Beth Israel Deaconess Medical Center Harvard Medical School Feinberg School of Medicine Harvard Medical School Boston, Massachusetts Northwestern Memorial Hospital Boston, Massachusetts Chicago, Illinois This activity is jointly sponsored by the University of Kentucky College of Medicine and CE Health Sciences Inc. New Insights Into the Diagnosis and Treatment of Single-Gene Disorders Associated With Cryptogenic Ischemic Stroke A Continuing Medical Education Monograph Editor Katherine B. Sims, MD Director, Developmental Neurogenetics Clinic Director, Neurogenetics DNA Diagnostic Laboratory Massachusetts General Hospital Associate Professor of Neurology Harvard Medical School Boston, Massachusetts Contributors Mark J. Alberts, MD, FAHA Professor of Neurology Section Chief, Stroke and Cerebrovascular Disease Northwestern University, Feinberg School of Medicine Director, Stroke Program Northwestern Memorial Hospital Chicago, Illinois Louis R. Caplan, MD Beth Israel Deaconess Medical Center Professor of Neurology Harvard Medical School Boston, Massachusetts To receive CME credit for this activity, please review the material in full and complete the online posttest and evaluation form at www.CECentral.com/getcredit (activity code MEN09182). A printable statement of credit will be issued upon successful completion of the required forms. 3 Accreditation This activity has been planned and implemented in accordance with the Essential Areas and Policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint sponsorship of the University of Kentucky College of Medicine and CE Health Sciences Inc. -
Why Do We Get New Families with Myotonic Dystrophy?
5 to 20 mutation 5 This mutation event probably only occurred once in human evolution in the shared common ancestor of 13 all Myotonic Dystrophy families. 11 12 14 15 20 to 35 20 repeats Repeats in this 5 to 15 repeats range are not 21 Repeats in this range associated with any are not associated symptoms and are 22 with any symptoms present at quite and are present at high frequency high frequency in the in the general 23 general population. population. They are They are genetically genetically unstable 24 very stable when when transmit- transmitted chang- ted, but increase in ing only very rarely. length quite slowly. 25 There is essen- There is definite tially zero risk of new risk of new Myo- 27 Myotonic Dystrophy tonic Dystrophy families arising from families arising from individuals with such individuals with such 30 repeats. repeats, but it may take many hundreds 33 of generations. 40 to 50 repeats 35 Repeats in this range are not associated with any symptoms, but are present at only very 40 low frequencies in the general population. They are though genetically unstable when transmitted, increasing in length very rapidly 45 and leading to new Myotonic Dystrophy families within a few generations. 50 60 to 3000 repeats 80 Repeats in this range are associated directly with Myotonic Dystrophy symptoms. The repeat is genetically very unstable and expands 300 rapidly in sucessive generations giving rise to the increased severity and decreased age of onset 1000 observed in Myotonic Dystrophy families. Myotonic Dystrophy Support Group Helpline 0115 987 0080 Myotonic dystrophy affects a wide range of body systems and varies dramatically in the relative severity of the symptoms and the age at which the first symptoms appear. -
Swallowing Diff Iculties in Myotonic Dystrophy
Swallowing Diff iculties in Myotonic Dystrophy by Jodi Allen Specialist Speech & Language Therapist, The National Hospital for Neurology & Neurosurgery, London Swallowing difficulties are an important aspect of Myotonic Dystrophy due to potentially serious complications. They should be identified early to help reduce risk of life-threatening complications. Management options often include swallowing strategies and sometimes alternative routes of feeding. These will be outlined as part of this booklet. Brief Summary • Myotonic Dystrophy can affect the muscles of your face, mouth and throat. • Weakness or stiffness (myotonia) in these muscles can cause problems with swallowing. • Swallowing problems can lead to weight loss and chest infections. • Problems can be identified early by looking out for signs and symptoms. These may include longer mealtimes, food sticking in the throat, needing to drink with meals and coughing and spluttering. Myotonic Dystrophy Support Group Helpline 0115 987 0080 • Swallowing problems should be assessed by a Speech and Language Therapist who can provide you with tailored advice and options. How do we normally eat and drink? The wind pipe (known as the trachea) and food pipe (known as the oesophagus) sit very close together in the throat, as shown below. The airway and food pipe sit close together in the throat When talking or engaging in an activity other than eating and drinking, the airway is open. This allows oxygen to pass into the lungs and expel waste gases out. At these times the entrance to the food pipe is closed. At regular intervals we initiate ‘a swallow.’ This allows saliva to move from the mouth into the food pipe.