Why Do We Get New Families with Myotonic Dystrophy?
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Genetic Background of Anticipation P Teisberg MD
JOURNAL OF THE ROYAL SOCIETY OF MEDICINE Volume 88 April 1995 The genetic background of anticipation P Teisberg MD J R Soc Med 1995;88:185-187 Keywords: genetics; anticipation; triplet repeats; neurological disorders Anticipation was controversial impairment and increased infant mortality were observed. Anticipation may be defined as the occurrence of a genetic The sequence of events often ends in congenital MD with its disorder at progressively earlier ages in successive severe clinical manifestation of mental retardation and generations. The disease moreover occurs with increasing muscular dystrophy. Later, clinical studies confirmed these severity. The concept emerged early in this century mainly observations and described a dominant inheritance pattern through descriptive dinical studies ofmyotonic dystrophy1'2. which could not be explained by classical Mendelian Later studies have added other disease entities to a list of mechanisms8. states showing anticipation, the most notable being Another phenomenon which did not fit easily into the Huntington's disease3. In one form of inherited mental concepts of genetics was the finding that congenital MD was retardation, the fragile X syndrome, the term 'the Sherman transmitted almost exclusively via affected mothers9. paradox' describes a very similar phenomenon4. In the fragile X syndrome, anticipation is manifested in a Towards the middle of this century, basic research in different manner. This is the most common cause of familial genetics had given us a much clearer understanding of mental retardation. It segregates in families as an X-linked Mendelian inheritance. It became increasingly difficult to dominant disorder with reduced penetrance. When reconcile the originally described phenomenon of chromosomes are stained a fragile site on the X anticipation with a concept of genes as stable elements chromosome may be seen in a proportion of cells taken only changed by the rare mutation. -

Original Articles Anticipation Resulting in Elimination of the Myotonic
J7 Med Genet 1994;31:595-601 595 Original articles J Med Genet: first published as 10.1136/jmg.31.8.595 on 1 August 1994. Downloaded from Anticipation resulting in elimination of the myotonic dystrophy gene: a follow up study of one extended family C E M de Die-Smulders, C J Howeler, J F Mirandolle, H G Brunner, V Hovers, H Bruggenwirth, H J M Smeets, J P M Geraedts Abstract muscular manifestations, it is characterised by We have re-examined an extended myo- multiple systemic effects including cataract, tonic dystrophy (DM) family, previously mental retardation, cardiac involvement, and described in 1955, in order to study the testicular atrophy. Extreme variability is one of long term effects of anticipation in DM the hallmarks of the disease; clinical studies and in particular the implications for have led to the recognition of four disease types families affected by this disease. This fol- on the basis of age at onset and core symptoms: low up study provides data on 35 gene late onset (mild) type, adult onset (classical) carriers and 46 asymptomatic at risk type, childhood, and congenital type.'"3 family members in five generations. Anticipation, increasing severity and earlier Clinical anticipation, defined as the cas- age at onset in successive generations, has been cade ofmild, adult, childhood, or congen- observed in DM since the beginning of this ital disease in subsequent generations, century, but remained unexplained and contro- appeared to be a relentless process, oc- versial until recently."- With the discovery of curring in all affected branches of the an unstable CTG trinucleotide repeat in the 3' family. -

Fact Sheet 54| FRAGILE X SYNDROME This Fact Sheet
11111 Fact Sheet 54| FRAGILE X SYNDROME This fact sheet describes the condition Fragile X and includes a discussion of the symptoms, causes and available testing. In summary Fragile X is a condition caused by a change in the FMR-1 gene, on the X chromosome It is characterised by particular physical features, varying degrees of learning difficulties and behavioural and emotional problems Fragile X affects around 1 in 4,000 males and between 1 in 5,000 and 1 in 8,000 females. WHAT IS FRAGILE X SYNDROME? WHAT CAUSES FRAGILE X SYNDROME? Fragile X syndrome is the most common known Fragile X syndrome is caused by a variation in the cause of inherited intellectual disability. genetic information in the FMR-1 gene. Genes are Intellectual problems in people with fragile X made up of a string of three letter ‘words’, or syndrome can vary from mild learning difficulties triplets, using the letters A,T, C & G. The FMR-1 through to severe intellectual disability. gene codes for a protein called FMRP that is necessary for usual brain development and/or Emotional and behavioural problems may also be function. In the FMR-1 gene, the triplet word present. ‘CGG’ can be repeated many times. When the Females with fragile X syndrome show varying number of ‘CGG’ repeats in the FMR-1 gene degrees of the condition, but are usually less increases over a critical number, the gene severely affected than males. becomes so long that it becomes faulty and the production of the FMRP is disrupted. The features of the condition, and their severity, are related to the genetic information in the faulty gene causing the condition. -

The Myotonic Dystrophies: Diagnosis and Management Chris Turner,1 David Hilton-Jones2
Review J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.2008.158261 on 22 February 2010. Downloaded from The myotonic dystrophies: diagnosis and management Chris Turner,1 David Hilton-Jones2 1Department of Neurology, ABSTRACT asymptomatic relatives as well as prenatal and National Hospital for Neurology There are currently two clinically and molecularly defined preimplantation diagnosis can also be performed.7 and Neurosurgery, London, UK 2Department of Clinical forms of myotonic dystrophy: (1) myotonic dystrophy Neurology, The Radcliffe type 1 (DM1), also known as ‘Steinert’s disease’; and Anticipation Infirmary, Oxford, UK (2) myotonic dystrophy type 2 (DM2), also known as DMPK alleles greater than 37 CTG repeats in length proximal myotonic myopathy. DM1 and DM2 are are unstable and may expand in length during meiosis Correspondence to progressive multisystem genetic disorders with several and mitosis. Children of a parent with DM1 may Dr C Turner, Department of Neurology, National Hospital for clinical and genetic features in common. DM1 is the most inherit repeat lengths considerably longer than those Neurology and Neurosurgery, common form of adult onset muscular dystrophy whereas present in the transmitting parent. This phenomenon Queen Square, London WC1N DM2 tends to have a milder phenotype with later onset of causes ‘anticipation’, which is the occurrence of 3BG, UK; symptoms and is rarer than DM1. This review will focus increasing disease severity and decreasing age of onset [email protected] on the clinical features, diagnosis and management of in successive generations. The presence of a larger Received 1 December 2008 DM1 and DM2 and will briefly discuss the recent repeat leads to earlier onset and more severe disease Accepted 18 December 2008 advances in the understanding of the molecular and causes the more severe phenotype of ‘congenital’ pathogenesis of these diseases with particular reference DM1 (figure 2).8 9 A child with congenital DM 1 to new treatments using gene therapy. -

X-Linked Myotubular Myopathy and Chylothorax
ARTICLE IN PRESS Neuromuscular Disorders xxx (2007) xxx–xxx www.elsevier.com/locate/nmd Case report X-linked myotubular myopathy and chylothorax Koenraad Smets * Department of Neonatology, Ghent University Hospital, De Pintelaan 185, B-9000 Ghent, Belgium Received 10 August 2007; received in revised form 4 October 2007; accepted 24 October 2007 Abstract X-linked myotubular myopathy usually presents at birth with hypotonia and respiratory distress. Phenotypic presentation, however, can be extreme variable. We report on a newborn baby, who presented with the severe form of the disease. In the second week of life, he developed a clinically relevant chylothorax, needing drainage and treatment with octreotide acetate. Pleural effusions are frequently described in patients with congenital myotonic dystrophy. To our knowledge, the association of chylothorax and X-linked myotubular myopathy has not been described to date. As chylothorax could not be attributed to any evident condition in this child, perhaps it may be added to the clinical spectrum of X-linked myotubular myopathy. Ó 2007 Elsevier B.V. All rights reserved. Keywords: X-linked myotubular myopathy; Chylothorax 1. Introduction drainage (Fig. 1). Laboratory examination was compatible with chylothorax (5230 white blood cells/ll, 98% lympho- Congenital myopathies often present with hypotonia cytes; chylomicrons were present; triglycerides 746 mg/dl). and respiratory distress from birth, although their expres- There were no central venous catheters in place who could sion may be delayed. In most cases muscle biopsy is war- have caused thrombosis, impairing lymphatic flow, neither ranted for definitive diagnosis. In some instances could any other risk factor for chylothorax be identified. -

Diverse Mechanisms of Trinucleotide Repeat Disorders: an Exploration of Fragile X Syndrome and Huntington’S Disease Cara Strobel
Undergraduate Review Volume 9 Article 30 2013 Diverse Mechanisms of Trinucleotide Repeat Disorders: An Exploration of Fragile X Syndrome and Huntington’s Disease Cara Strobel Follow this and additional works at: http://vc.bridgew.edu/undergrad_rev Part of the Cell Biology Commons Recommended Citation Strobel, Cara (2013). Diverse Mechanisms of Trinucleotide Repeat Disorders: An Exploration of Fragile X Syndrome and Huntington’s Disease. Undergraduate Review, 9, 151-156. Available at: http://vc.bridgew.edu/undergrad_rev/vol9/iss1/30 This item is available as part of Virtual Commons, the open-access institutional repository of Bridgewater State University, Bridgewater, Massachusetts. Copyright © 2013 Cara Strobel Diverse Mechanisms of Trinucleotide Repeat Disorders: An Exploration of Fragile X Syndrome and Huntington’s Disease CARA STROBEL Cara Strobel authored this essay for the Cell Biology course in the spring semester of 2012. Given free rinucleotide repeat disorders are an umbrella group of genetic diseases reign with a cell biology related topic, that have been well described clinically for a long time; however, the she wanted to explore and contrast scientific community is only beginning to understand their molecular the specifics of several prevalent basis. They are classified in two basic groups depending on the location Tof the relevant triplet repeats in a coding or a non-coding region of the genome. disorders. Cara plans to apply to Repeat expansion past a disease-specific threshold results in molecular and cellular medical school in Spring 2013. abnormalities that manifest themselves as disease symptoms. Repeat expansion is postulated to occur via slippage during DNA replication and/or transcription- mediated DNA repair. -

Relationship Between C9orf72 Repeat Size and Clinical Phenotype
Available online at www.sciencedirect.com ScienceDirect Relationship between C9orf72 repeat size and clinical phenotype 1,2,3,4 1,2 1,2 Sara Van Mossevelde , Julie van der Zee , Marc Cruts 1,2 and Christine Van Broeckhoven Patient carriers of a C9orf72 repeat expansion exhibit Patients carrying a C9orf72 repeat expansion are remark- remarkable heterogeneous clinical and pathological able heterogeneous in clinical presentation, not only characteristics suggesting the presence of modifying factors. between families but also within families [3,4 ]. The In accordance with other repeat expansion diseases, repeat majority of the expansion carriers present clinically with length is the prime candidate as a genetic modifier. FTD and/or ALS. 73–100% of C9orf72 FTD patients Observations of earlier onset ages in younger generations of exhibit the behavioral variant (bvFTD) [5–15]. In C9orf72 large families suggested a mechanism of disease anticipation. ALS patients, the relative frequency of a bulbar symptom Yet, studies of repeat size and onset age have led to conflicting onset (29–89% [5,9,11,13,15–18]) is higher than in ALS results. Also, the correlation between repeat size and diagnosis patients without the expansion [13,16–18]. Apart from is poorly understood. We review what has been published FTD and ALS, several other clinical diagnoses have been regarding C9orf72 repeat size as modifier for phenotypic described (Figure 1) [9,19–31]. Parkinsonism is fre- characteristics. Conclusive evidence is lacking, partly due to quently reported in C9orf72 repeat expansion carriers, the difficulties in accurately defining the exact repeat size and but the expansion does not seem to be associated with the presence of repeat variability due to somatic mosaicism. -

The Natural History of Machado-Joseph Disease an Analysis of 138 Personally Examined Cases A
THE CANADIAN JOURNAL OF NEUROLOGICAL SCIENCES QUEBEC COOPERATIVE STUDY OF FRIEDREICH'S ATAXIA The Natural History of Machado-Joseph Disease An analysis of 138 personally examined cases A. Barbeau, M. Roy, L. Cunha, A.N. de Vincente, R.N. Rosenberg, W.L. Nyhan, P.L. MacLeod, G. Chazot, L.B. Langston, D.M. Dawson and P. Coutinho ABSTRACT: We have examined 138 cases of a disorder previously described in people of Portuguese origin and which has received many names. By computer analysis of 46 different items of a standardized neurological examination carried out in each patient, we have been able to delineate the main components of the clinical presentation, to conclude that the marked variability in clinical expressions does not negate the homogeneity of the disorder, and to describe the natural history of this entity which should be called, for historical reasons, "Machado-Joseph Disease". This hereditary disease has an autosomal dominant pattern of inheritance, presenting as a progressive ataxia with external ophthalmoplegia, and should be classified within the group of "Ataxic multisystem degenerations". When the disease starts before the age of 20, it may present with marked spasticity, of a non progressive nature but often so severe that it can be accompanied by "Gegenhalten" countermovements and dystonic postures but little frank dystonia. There are few true extrapyramidal symptoms except akinesia. When the disease starts after the age of 50, the clinical spectrum is mostly that of an amyotrophic polyneuropathy with fasciculations accompanying the ataxia. For all the other cases the clinical picture is a c.ontinuum between these two extremes, the main determinant of the clinical phenotype being the age of onset and a secondary factor, the place of origin of the given kindred. -

Patterns of Conservation of Spliceosomal Intron Structures and Spliceosome Divergence in Representatives of the Diplomonad and Parabasalid Lineages Andrew J
Hudson et al. BMC Evolutionary Biology (2019) 19:162 https://doi.org/10.1186/s12862-019-1488-y RESEARCH ARTICLE Open Access Patterns of conservation of spliceosomal intron structures and spliceosome divergence in representatives of the diplomonad and parabasalid lineages Andrew J. Hudson1,2†, David C. McWatters1,2†, Bradley A. Bowser3, Ashley N. Moore1,2, Graham E. Larue3, Scott W. Roy3,4 and Anthony G. Russell1,2* Abstract Background: Two spliceosomal intron types co-exist in eukaryotic precursor mRNAs and are excised by distinct U2- dependent and U12-dependent spliceosomes. In the diplomonad Giardia lamblia, small nuclear (sn) RNAs show hybrid characteristics of U2- and U12-dependent spliceosomal snRNAs and 5 of 11 identified remaining spliceosomal introns are trans-spliced. It is unknown whether unusual intron and spliceosome features are conserved in other diplomonads. Results: We have identified spliceosomal introns, snRNAs and proteins from two additional diplomonads for which genome information is currently available, Spironucleus vortens and Spironucleus salmonicida, as well as relatives, including 6 verified cis-spliceosomal introns in S. vortens. Intron splicing signals are mostly conserved between the Spironucleus species and G. lamblia. Similar to ‘long’ G. lamblia introns, RNA secondary structural potential is evident for ‘long’ (> 50 nt) Spironucleus introns as well as introns identified in the parabasalid Trichomonas vaginalis. Base pairing within these introns is predicted to constrain spatial distances between splice junctions to similar distances seen in the shorter and uniformly-sized introns in these organisms. We find that several remaining Spironucleus spliceosomal introns are ancient. We identified a candidate U2 snRNA from S. vortens, and U2 and U5 snRNAs in S. -
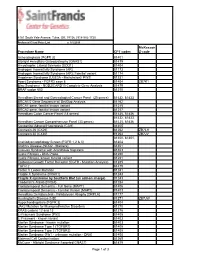
Procedure Name CPT Codes Mckesson Z-Code Achondroplasia
6161 South Yale Avenue, Tulsa, OK, 74136 | 918-502-1720 Preferred Client Price List v. 1/1/2019 McKesson Procedure Name CPT codes Z-code Achondroplasia {FGFR 3} 81401 Albright Hereditary Osteodystrophy {GNAS1} 81479 Amyotrophic Lateral Sclerosis {SOD1} 81404 Androgen Insensitivity Syndrome {AR} 81173 Androgen Insensitivity Syndrome {AR}; Familial variant 81174 Angelman Syndrome {UBE3A - Methylation}/ PWS 81331 Apert Syndrome - FGFR2 exon 8 81404 ZB7K1 Blau Syndrome - NOD2/CARD15 Complete Gene Analysis 81479 BRAF codon 600 81210 Hereditary Breast and Gynecological Cancer Panel (25 genes) 81432, 81433 BRCA1/2 Gene Sequence w/ Del/Dup Analysis 81162 BRCA1 gene, familial known variant 81215 BRCA2 gene, familial known variant 81217 Hereditary Colon Cancer Panel (18 genes) 81435, 81436 81432, 81433, Hereditary Cancer Comprehensive Panel (33 genes) 81435, 81436 Congenital Adrenal Hyperplasia {CAH} 81405 Connexin 26 {CX26} 81252 ZB7LH Connexin 30 {CX30} 81254 ZB7JV 81400, 81401, Craniodysmorphology Screen {FGFR 1,2 & 3} 81404 Crohn's Disease {NOD2 - Markers} 81401 Crouzon Syndrome with Acanthosis Nigricans 81403 Cystic Fibrosis - DNA Probe 81220 Cystic Fibrosis, known familial variant 81221 Epidermal Growth Factor Receptor {EGFR - Mutation Analysis} 81235 FGFR 2 81479 Factor V Leiden Mutation 81241 Fragile X Syndrome {FRAX1} 81243 Fragile X syndrome by Southern Blot (an add-on charge) 81243 Friederich's Ataxia {FRDA} 81284 Frontotemporal Dementia - Full Gene (MAPT) 81406 Frontotemporal Dementia - Familial Variant (MAPT) 81403 Hereditary Dentatorubral -
Swallowing Diff Iculties in Myotonic Dystrophy
Swallowing Diff iculties in Myotonic Dystrophy by Jodi Allen Specialist Speech & Language Therapist, The National Hospital for Neurology & Neurosurgery, London Swallowing difficulties are an important aspect of Myotonic Dystrophy due to potentially serious complications. They should be identified early to help reduce risk of life-threatening complications. Management options often include swallowing strategies and sometimes alternative routes of feeding. These will be outlined as part of this booklet. Brief Summary • Myotonic Dystrophy can affect the muscles of your face, mouth and throat. • Weakness or stiffness (myotonia) in these muscles can cause problems with swallowing. • Swallowing problems can lead to weight loss and chest infections. • Problems can be identified early by looking out for signs and symptoms. These may include longer mealtimes, food sticking in the throat, needing to drink with meals and coughing and spluttering. Myotonic Dystrophy Support Group Helpline 0115 987 0080 • Swallowing problems should be assessed by a Speech and Language Therapist who can provide you with tailored advice and options. How do we normally eat and drink? The wind pipe (known as the trachea) and food pipe (known as the oesophagus) sit very close together in the throat, as shown below. The airway and food pipe sit close together in the throat When talking or engaging in an activity other than eating and drinking, the airway is open. This allows oxygen to pass into the lungs and expel waste gases out. At these times the entrance to the food pipe is closed. At regular intervals we initiate ‘a swallow.’ This allows saliva to move from the mouth into the food pipe. -

Beyond Type 1 and Type 2 Diabetes; Why Correct Diabetes Classification Is Important Gabriel I. Uwaifo, MD Dept of Endocrinology
Beyond Type 1 and Type 2 Diabetes; Why Correct Diabetes Classification is Important Gabriel I. Uwaifo, MD, FACP, FTOS, FACE. Dept of Endocrinology, Diabetes, Metabolism and Weight Management, Ochsner Medical Center Objectives To highlight various classification methods of diabetes To highlight the importance and consequences of appropriate diabetes classification To provide suggested processes for diabetes classification in primary care settings and indices for specialty referral Presentation outline 1. Case presentations 2. Diabetes classification; past present and future 3. Diabetes classification; why is it important? 4. Suggested schemas for diabetes classification 5. Case presentation conclusions 6. Summary points and conclusions 3 Demonstrative cases Patient DL is a 56 yr old AA gentleman with a BMI of 24 referred for management of his “type 2 diabetes”. He is on basal bolus insulin with current HBA1c of 8.3. His greatest concern is on account of recent onset progressive neurologic symptoms and gaite unsteadiness Patient CY is a 21 yr old Caucasian lady with BMI of 28 and strong family history of diabetes referred for management of her “type 2 diabetes”. She is unsure if she even has diabetes as she indicates most of the SMBGs are under 160 and her current HBA1 is 6.4 on low dose metformin. Patient DR is a 54 yr old Asian lady with BMI of 36 and long standing “type 2 diabetes”. She has been referred because of poor diabetes control on multiple oral antidiabetics and persistent severe hypertriglyceridemia. Questions; Do all