Handbook-Of-Ferroalloys.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rules and Options
Rules and Options The author has attempted to draw as much as possible from the guidelines provided in the 5th edition Players Handbooks and Dungeon Master's Guide. Statistics for weapons listed in the Dungeon Master's Guide were used to develop the damage scales used in this book. Interestingly, these scales correspond fairly well with the values listed in the d20 Modern books. Game masters should feel free to modify any of the statistics or optional rules in this book as necessary. It is important to remember that Dungeons and Dragons abstracts combat to a degree, and does so more than many other game systems, in the name of playability. For this reason, the subtle differences that exist between many firearms will often drop below what might be called a "horizon of granularity." In D&D, for example, two pistols that real world shooters could spend hours discussing, debating how a few extra ounces of weight or different barrel lengths might affect accuracy, or how different kinds of ammunition (soft-nosed, armor-piercing, etc.) might affect damage, may be, in game terms, almost identical. This is neither good nor bad; it is just the way Dungeons and Dragons handles such things. Who can use firearms? Firearms are assumed to be martial ranged weapons. Characters from worlds where firearms are common and who can use martial ranged weapons will be proficient in them. Anyone else will have to train to gain proficiency— the specifics are left to individual game masters. Optionally, the game master may also allow characters with individual weapon proficiencies to trade one proficiency for an equivalent one at the time of character creation (e.g., monks can trade shortswords for one specific martial melee weapon like a war scythe, rogues can trade hand crossbows for one kind of firearm like a Glock 17 pistol, etc.). -

Parang Ilang Sebagai Interpretasi Falsafah Alam Takambang Jadi Guru Dalam Budaya Masyarakat Iban
ASIAN PEOPLE JOURNAL 2020, VOL 3(1), 1-18 e-ISSN: 2600-8971 ASIAN PEOPLE JOURNAL, 2020, VOL 3(1), 1-18 https://doi.org/10.37231/apj.2020.3.1.118 https://journal.unisza.edu.my/apj PARANG ILANG SEBAGAI INTERPRETASI FALSAFAH ALAM TAKAMBANG JADI GURU DALAM BUDAYA MASYARAKAT IBAN (Parang Ilang As An Interpretation Of Alam Takambang Jadi Guru Philosophy In Iban’s Culture) Adilawati Asri1*, Noria Tugang1 1Fakulti Seni Gunaan dan Kreatif, Universiti Malaysia Sarawak, 94300 Kota Samarahan, Sarawak, Malaysia *Corresponding author: [email protected] Received: 5 September 2019 • Accepted: 25 November 2019 • Published: 30 April 2020 Abstract This research explores the craft and creativity of the art of Ilang as an interpretation of the philosophy of ‘Being a Teacher’ in the life of the Iban community. Symbolism towards the elements of nature is often used to express various ideas and meanings about their practices, culture, and life. This research uses a qualitative approach as a whole. Data obtained through qualitative approach using observations and interviews. Overall research has found that the human mind is made up of natural properties. Humans learn from nature and create the aesthetics they learn from it. Nature is made as a 'teacher', a human mind built from nature. This means that the developing world teaches humans to think creatively and to create something using natural resources, such as parang Ilang. Every event that happens around human life may not be separated from nature. The Iban people live in a natural environment. For them, nature is a ‘teacher’ to solve all the problems that occur in their lives. -
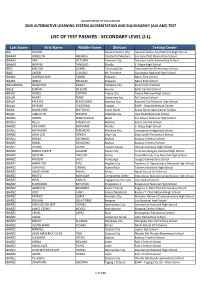
List of Test Passers - Secondary Level (I-L)
DEPARTMENT OF EDUCATION 2015 ALTERNATIVE LEARNING SYSTEM ACCREDITATION AND EQUIVALENCY (ALS A&E) TEST LIST OF TEST PASSERS - SECONDARY LEVEL (I-L) Last Name First Name Middle Name Division Testing Center IBA JHUGIN General Santos City General Santos City National High School IBABAO WENELYN ABENOJA Occidental Mindoro San Jose Pilot Elementary School IBABAO ARA VICTORIA Tanauan City Tanauan South Elementary School IBABAO NORVIN TANGLAO Manila V. Mapa High School IBAD MELODY GUINID Tacurong City Tacurong Pilot Elementary School IBAG SADEN CLAUDIO Mt. Province Guinzadan National High School IBAJAN VANESSA MAE TABON Palawan Narra Pilot School IBAJAN GERLIE ARCILLAS Palawan Narra Pilot School IBALARROSA GENEVIEVE RAGOT Tacloban City Rizal Central School IBALE EDRIAN DE LEON Aurora Baler Central School IBALIG AGNES CONTAD Tanjay City Tanjay National High School IBALIN HENRY PEREZ Camarines Sur Pili Central School IBALLA PAULINE BONGCAWEL Bacolod City Bacolod City National High School IBALLA REYMAR CONSIGNA Siargao BJMP - Dapa Detention Center IBANA DIANA ROSE MATOCDO Ilocos Norte Ilocos Norte Agricultural College IBANA CARLOS JR. PONAYO Calamba City Jose Rizal Memorial School IBAÑES ARDEN MANTIQUILLA Bohol San Roque National High School IBAÑES NELIA INTERVALO Batanes Basco Central School IBAÑEZ JON MARK ABRAHAM Manila V. Mapa High School IBAÑEZ RHAYMARK PEÑAROYO Marikina City Concepcion Integrated School IBAÑEZ JOHN LEO SONDIA Silay City Silay South Elementary School IBAÑEZ ROGER LUCHEROS Baybay Baybay I Central School IBAÑEZ RODEL ESCASINAS Baybay Baybay I Central School IBAÑEZ ALVINA ACASO Eastern Samar Dolores National High School IBAÑEZ MARIA JOSIEFE SALAZAR Davao City Francisco Bangoy National High School IBAÑEZ JOESTEN LIBO-ON Cavite Trece Martires City National High School IBAÑEZ RHOJUN LALIC Pampanga Guagua Elementary School IBAÑEZ MARIE PAZ INTO Angeles City Francisco G. -

12 Disember 2001
'X ♦» UAt&WfA 9AAN PETIKAN TTTAH seUAOA dan aaniaara- hOALAM amatan barpuaaa, ^ kasKi bag* ewwa* Kita bukan sa}a diamal dan t^aiats AM^aoar iaMw d< diiattti untuk merahan did -r>T—----" ■ j ari melafcukan dari nsart se'sv,^^ twataw iv- arlura-perkara yang di- Anr acAan Ramadan.* kegah malahan juga dl- =et*a8art SaAa*- lanlurtcan untuk aentlaaa Ki^rs Weaaflaae yirvg icart kacamertangan da- apa {uga bldang dan fartanynkan o*ah Ai-^ m yang tdts lakuKan." • cnxri dafi A/sy^ Raoa^ aampana Hari Raya ihutetii- Hri200a BRUNEI TAHUN 46 BILANCAN 50 RABU 12 DISEMBER. 2001/1422 ^vOWaaj 26 jjlj DITERBITKAN PAOA SETIAP HARI RABU PERCUMA )UN]UNG ZIARAH HARI RAYA AIDILFITR! YANG 1422 HIJRAH NARIK MENJUNJUNG lilah Kebawah Duli Yang Maha Muliu Paduka Scri Baginda Sultan Haji Hassanal uan: Bolkiah Mu'izzaddin Waddaulah, Sultan dan Yang Oi-Pcrtuan Negara Brunei Darussaiont. Yang Amai Muiia Pengiran Penggawa Laita SentaiB Istiadai Pelbagai prodiik dan Diraja Dalam Istana, Grand Chamberlain meng- perkhidmatan umumkan bahawa sempena Hari Raya Aidilfitri Thhun 1422 Hijrah, Istana Nuiul Iman akan dibuka - Muka 2 selama tiga hari. mulai dari Hari Raya Kedua dari jam 10.00 pagi hingga 12.00 tengah han dan dari jam 2.00 petang hingga 4.00 petang. bagi membolehkan elirik Masa Silam : orang ramai mengadap dan menziarahi Kebawah Duli Yang Maha Mulia Paduka Seri Baginda Sultan Terkenang tempat dan Yang Di-Pertuan Negara Brunei Darussalom dan kerabat dtraja. jatuh • Muka 3 Penglihatan ■Mempelbagaikan KPaawAH Dull Yang Maha Mulls Paduka Serl Baginda Sultan Haji Hassanal Bolkiah Mu'izzaddin Waddaulah, Sultan dan lekonomi menjadi Vang CM-Partuan Negara Brunei Darussalam ketika berkenan menerlma mengadap dua setiausaha tetap yang baru dllantlk laitu Awang Ha)l DIrIa, Setiausaha Tetap Kementerlan Perindustrlan dan Sumber-Sumber Utama dan Oslo Paduka Awang Ikeperluan utama Hilal Syawal HaJI Dtnl, Setiausaha Tetap Kementerlan tCrttudayaan, Bella dan Sukan. -

Kamus Minangkabau-Indonesia
Kamus Minangkabau-Indonesia Oleh: Marah Rusmali Amir Hakim Usman Syahwin Nikelas Nurzuir Husin Busri Agusli Lana M.Yamin ; Isna Sulastri Irfani Basri Pusat Pembinaan dan Pengembangan Bahasa Departemen Pendidikan dan Kebudayaan Jakarta 1985 an Penyunting Hans Lapal Sen : K---85.031 Cetakan Pertama Naskah buku mi, yang semula merupakan hasil Proyek Pengembangan Bahasa dan Sastra Indonesia dan Daerah tahun 1982/1983, diterbitkan dengan dana Proyek Pengembangan Bahasa dan Sastra Indonesia. Staf Intl Proyek Drs. Tony S. Rachmadie (Pemimpin), Samidjo (Bendaharawan), Drs. S.R.H. Sitanggang (Sekretaris), Drs. S. Amran Tasai, Drs. A. Patoni, Dra. Siti Zahra Yundiafi, dan Drs. E. Zainal Arifin (Asisten). Sebagian atau seluruh isi buku mi dilarang diperbanyak dalam bentuk apa pun tanpa izin tertulis dari penerbit, kecuali dalam hal kutipan untuk ke- perluan penulisan artikel atau karangan ilmiah. Alamat Penerbit Pusat Pembinaan dan Pengembangan Bahasa Jalan Daksinapati Barat IV, Rawamangun Jakarta 13220 PRAKATA Sejak Rencana Pembangunan Lima Tahun II (1974), telah digariskan kebijakan pembinaan dn pengembangan kebudayaan nasional dalam ber. bagal seginya. Dalam garis haluan mi, masalah kebahasaan merupakan salah satu masalah kebudayaan nasional yang perlu digarap dengan sungguh. sungguh dan berencana sehingga tujuan akhir pembinaan dan pengembangan bahasa Indonesia dan daerah, termasuk sastranya, dapat tercapai. Tujuan akhir pembinaan dan pengembangan itu, antara lain, adalah meningatkan mutu kemampuan menggunakan bahasa Indonesia sehingga sarana -

Depuration Plant Owner Defies State
County planners cut off Silverside extension, B1 GREATER RED BANK EATONTOWN Burned up Cops a plea LONG BRANCH White Sox1 Britt Burns Death penalty witness guilty of armed robbery. Today's Forecast: smokes Yankees, 1-0 Sunny today, warmer tomorrow Page B3 Page B1 Complete weather on A2 The Daily Register VOL.106 NO. 38 YOUR HOMETOWN NEWSPAPER . SINCE 1878 TUESDAY, AUGUST 16, 1983 . 25 CENTS Depuration plant owner defies state By RAY GERMANN tion boarded Harvey's boat Satur- "The enforcement people will "I'm tired of being pooped since June 1, and has changed areas clammers — who are primarily day, and informed him he was en- decide what summons, or sum- upon," he declared. every few weeks. from Ocean County — can now State officials are pondering couraging illegal clamming, accord- .nonses, his actions warrant," she Before clamming began June 1, Officials of the Bureau of harvest on Monday, in addition to whether to take legal action against ing to Gale Critchlow, spokesperson said. The DEP's marine police state officials agreed to allow Shellfisheries and the DEP's Wednesday and Friday. Highlands clam depuration plant for the DEP's Bureau of planned to discuss the incident with Harvey to harvest the area of his Division of Fish. Game and Wildlife Under the relay program, owner Jayson Harvey after he de- Shellfisheries. DEP officials before taking action, choosing, Harvey said. "I believe I voted Friday to expand the Bayshore clams are transported to fied state guidelines and started said Robert Winkel, a DEP enforce- Bayshore hard-clam relay program cleaner Ocean County waters and A "buy boat" owned by Harvey, should have that choice, and that's clamming in restricted areas of the ment officer. -

Enciclopedia Delle Illustrata Delle Armi Bianche
Enciclopedia delle illustrata delle armi bianche Abbasi Versione del Rajput della spada lunga indiana con lama rinforzata da nastri di metallo perforato. La Abbasai Talwar è una sciabola del Punjab. Accetta (vedi anche Scure); (Ing. Battle-axe; fr. Hache d'armes; ted. Streitaxt) Piccola scure molto in uso in Europa centro-orientale nel 1500-1600. A e B: Accetta; C e D: Scure d'arme; E: Accetta alla siciliana; F: Steigerhacke; G: Scure del guastatore. L’accetta alla siciliana è molto lavorata ed era arma cerimoniale nel 1600-1700. La Steigerhacke era arma cerimoniale dei minatori tedeschi e svedesi dal 150 al 1700. (Immagine di C. De Vita) 1 Enciclopedia delle illustrata delle armi bianche Achico Tipo di bolas con tre palle. Acinaces (meglio Akinakes) Corta spada persiana (500 a. Cr.) originaria degli Sciti (ricostruzione di fantasia). Aclys Corto giavellotto romano Adarga Arma che pare fosse usata dai nel 1400 dagli arabi; era formata da uno scudo con una lama infissa perpendicolarmente su di esso e dall’impugnatura formata da due 2 Enciclopedia delle illustrata delle armi bianche lance contrapposte. Agny Astra Specie di razzo incendiario lanciato da un tubo di bambù, in uso presso gli antichissimi Hindu. Ahir Corta spada ricurva dei Mahratta (immagine di una riproduzione di fantasia). Ahlspiess Quadrellone munito di ampio disco di arresto alla base del ferro; in uso in ambiente tedesco nel 1400. Aikuchi, Kusungobu La parola aikuchi indica una particolare montatura del coltello giapponese, senza guardia con lama lunga 0,95 piedi (cm. 27,5 circa) e di solito senza nastratura con in vista la pelle di razza e le borchie (menuki). -

May 13 - 19, 2018
MAY 13 - 19, 2018 staradvertiser.com GROWING GIRLS The March sisters are back with a joint PBS and BBC miniseries production of Louisa May Alcott’s Little Women. Meg (Willa Fitzgerald), Jo (Maya Hawke), Beth (Annes Elwy) and Amy (Kathryn Newton) grow together in this authentic adaptation, hoping to empower a new generation of girls. Premieres Sunday, May 13, on PBS as part of Masterpiece Classic. NEW EPISODE! Join host, Lyla Berg as she sits down with guests Meet the who share their work on moving our community forward. people SPECIAL GUESTS INCLUDE: and places Conrad Nonaka, Director, KCC Culinary Institute of the Pacific that make 1st & 3rd Natalie McKinney, Executive Director, Kokua Hawaii Foundation Hawai‘i Wednesday of the Month, Bob Hampton, Chairman, Waikiki Beach Activities Robin Kumabe Executive Director, Touch a Heart Hawaii 6:30 pm | Channel 53 , olelo.org special. Donne Dawson, Hawaii State Film Commissioner ON THE COVER | LITTLE WOMEN Splendid sisters PBS breathes new life into Louisa less but also fatherless, as the patriarch has may be in large part due to the success of her May Alcott’s ‘Little Women’ been called off to war. Left to fend for them- parents. The actress is the daughter of Uma selves, the March women are eager to prove Thurman (“Kill Bill: Vol. 1,” 2003) and Ethan that they’re more than capable of doing so. Hawke (“Training Day,” 2001), two beloved ac- By K.A. Taylor Each sister is undeniably a March, yet entirely tors whose passion for the craft seems to be TV Media unique. -

Kata Pengantar
KATA PENGANTAR Berkat Rahmat Allah Yang Maha Pengasih lagi Maha Penyayang, kami telah menyelesaikan Buku Data Statistik Sektoral dan Informasi Geospasial Tematik Bidang Lingkungan Hidup dan Kehutanan, yang merupakan bagian dari Kegiatan Penyediaan Data Pembangunan Sektoral. Data dan informasi yang disajikan dalam Buku ini bersumber dari seluruh bidang lingkup Dinas Lingkungan Hidup dan Kehutanan Provinsi Banten. Kami mengucapkan terimakasih dan penghargaan kepada semua pihak yang telah membantu dalam penyusunan Buku Data Statistik Sektoral dan Informasi Geospasial Tematik Bidang Lingkungan Hidup dan Kehutanan Tahun 2018 ini. Kami menyadari masih adanya berbagai kekurangan dalam Buku Data Statistik Sektoral dan Informasi Geospasial Tematik Bidang Lingkungan Hidup dan Kehutanan Tahun 2018 ini, oleh karena itu saran dan kritik yang sifatnya membangun sangat dinantikan. Semoga Buku ini dapat memberikan manfaat bagi yang memerlukannya. Serang, November 2018 Kepala Dinas Lingkungan Hidup dan Kehutanan Provinsi Banten, Ir. H.M. Husni Hasan,CES Pembina Utama Madya / IV.d NIP. 19621222 199003 1 004 DATA STATISTIK SEKTORAL DAN INFORMASI i GEOSPASIAL TEMATIK BIDANG LINGKUNGAN HIDUP DAFTAR ISI Kata Pengantar .................................................................................................. i Daftar Isi ............................................................................................................. ii I. KONDISI UMUM WILAYAH ..................................................................... 1 A. Administratif, Luas -

November 12 - 18, 2017
NOVEMBER 12 - 18, 2017 staradvertiser.com SINNER TO SAINT In spite of his selfi sh past — or perhaps because of it — Kevin Finn (Jason Ritter) sets out to make the world a better place in Kevin (Probably) Saves the World. All the while, a celestial being known as Yvette (Kimberly Hébert Gregory) guides him on his mission. Airing Tuesday, Nov. 14, on ABC. NEW SHOW Join host, Lyla Berg as she sits down with guests Meet the who share their work on moving our community forward. WEDNESDAY! people SPECIAL GUESTS INCLUDE: and places Senator Stanley Chang Georja Skinner, Creative Industries Chief Officer, DBEDT that make Darryl Vincent, COO, U.S. Vets 1st & 3rd Wednesday Kimi Sakuda Takazawa, Director of Philanthropy, Boy Scouts of America Hawai‘i of the Month, 6:30 pm Channel 53 Daniel Alexander, Advocacy Planning olelo.org special. and Communication Director, Hawaii Bicycling League ON THE COVER | KEVIN (PROBABLY) SAVES THE WORLD TV with soul New ABC drama follows a “It’s about making ourselves better, and She may be hard on Kevin, but he seems reaching out and making the world better,” the to need some tough love; he hasn’t always man on a mission actress said. behaved so saintly. As Ritter expressed in a That’s exactly what Kevin is tasked with do- promo, Kevin “certainly wouldn’t seem like By Kyla Brewer ing, and he has an uphill climb. After hitting rock anyone’s first choice to save the world” — TV Media bottom, he returns to his childhood home to but that may very well be the point. -
Nahant ROCKS Tests to the Test from A1 TESTS MCAS, He Said
THURSDAY, NOVEMBER 16, 2017 Budget shortfall leaves Lynn feeling spent By Thomas Grillo “If we are unable to set the tax request of the Legislature is not money hasn’t reached the city’s recommended the city not is- ITEM STAFF rate and mail out the third quar- unusual, neither could recall the coffers. sue raises after union contracts ter tax bills by the end of the last time the city needed the ex- The trash fee is expected to expire, freeze hiring, contract LYNN — The city’s budget year, the city will not receive the emption. generate about $2 million annu- EMS services to a private com- de cit, now set at $5 million, $31 million in real estate taxes “It hasn’t happened in my ally, the meals tax should raise pany, and eliminate three dozen could become a $36 million prob- by Feb. 1 and we will be unable time on the job and I’ve been $700,000 and the two medical jobs. lem for Mayor-elect Thomas M. McGee if the city is unable to set to meet payroll,” Caron told the here since 1998,” said Caron. marijuana clinics slated to open Mayor Judith Flanagan Ken- the tax rate by year’s end. City Council Tuesday. “Without “Getting approval to send the in the city could raise as much nedy, who recently lost her bid Peter Caron, the city’s chief the Legislature’s approval, we tax bills is not a big deal, but the as $400,000 per year. But so far for a third term, said Caron is nancial of cer, wants the Leg- would have to borrow a signi - budget de cit is.” the city has not seen the cash. -

Kamus Lampungandonesia
TIDAK DIPERDAGANGKAN (JNTUKUMUM Kamus LampungAndonesia DATA en Kamus Lam pung Indonesia OIeh: Junaiyah H.M. dkk. Pusat Pembinaan dan Pengembangan Bahasa Departemen Pendidikan dan Kebudayaan Jakarta 1985 lffl Hak Cipta pada Departemen Pendidikan dan Kebudayaan SAW b is %mull, I : 1729 O 1101 L I...411 1 1t -- Cetakan Pertatna Naskah buku ml semula merupakan hasil Proyek Pengembangan Bahasa dan Sastra Indonesia dan Daerah—Jakarta 1976/1977 dlterbitkan dengan dana Proyek Pengembangan Bahasa dan Sastra Indonesia. Staf inti ProyekDrs. Hans Lapoliwa, M. Phil. (Pemimpin), Yusnan Yunus (Bendaharawan), Drs. Nafron Hasjim, Drs. Dendy Sugono (Sekretaris), Drs. Farid Hadi, Drs. S.R.H. Sitanggang, Drs. Tony S. Racmadie Drs. S. Amran TasaiDrs. A. Patoni, H. Abd. Mutalib, BA. (para asisten). Sebagian atau seluruh isi buku ml dilarang dipergunakan atau diperbanyak dalain bentuk apa pun tanpa izin tertulis daii penerbit kecuall dalam hal ku- tipan untuk keperluan penulisan artikel atau karangan ilmiah. Alamat penerbit: Pusat Pembinaan dan Pengembangan Bahasa Jalan Daksinapatl Barat IV, Rawamangun Jakarta Tirnur 13220. Iv PRAKATA Sejak Rencana Pembangunan Lima Tahun 11 (1974), telah digariskan kebi- jakan pembinaan dan pengembangan kebudayaan nasional dalam berbagai seginya. Dalam gris haluan mi, masalah kebahasaan dan kesastraan merupa- kan salah satu masalah kebuyaan nasional yang perlu digarap dengan sung- guh-sungguh clan berencana sehingga tujuan akhir pembinaan dan pengemba- ngan bahasa Indonesia dan daerah, termasuk sastranya, dapat tercapai. Tujuan