1 a Novel Genetic and Morphologic Phenotype of ARID2-Mediated Myelodysplasia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mir-376C Promotes Carcinogenesis and Serves As a Plasma Marker for Gastric Carcinoma
RESEARCH ARTICLE miR-376c promotes carcinogenesis and serves as a plasma marker for gastric carcinoma Pei-Shih Hung1, Chin-Yau Chen2, Wei-Ting Chen2, Chen-Yu Kuo3, Wen-Liang Fang4,5, Kuo-Hung Huang4,5, Peng-Chih Chiu5, Su-Shun Lo2,6* 1 Department of Education and Medical Research, National Yang-Ming University Hospital, Yilan, Taiwan, 2 Department of Surgery, National Yang-Ming University Hospital, Yilan, Taiwan, 3 Department of Medicine, National Yang-Ming University Hospital, Yilan, Taiwan, 4 Division of General Surgery, Veterans General Hospital±Taipei, Taipei, Taiwan, 5 Department of Dentistry, National Yang-Ming University Hospital, Yilan, Taiwan, 6 School of Medicine, National Yang-Ming University, Taipei, Taiwan a1111111111 [email protected] a1111111111 * a1111111111 a1111111111 a1111111111 Abstract Gastric carcinoma is highly prevalent throughout the world. Understanding the pathogenesis of this disease will benefit diagnosis and resolution. Studies show that miRNAs are involved in the tumorigenesis of gastric carcinoma. An initial screening followed by subsequent vali- OPEN ACCESS dation identified that miR-376c is up-regulated in gastric carcinoma tissue and the plasma Citation: Hung P-S, Chen C-Y, Chen W-T, Kuo C-Y, of patients with the disease. In addition, the urinary level of miR-376c is also significantly Fang W-L, Huang K-H, et al. (2017) miR-376c increased in gastric carcinoma patients. The plasma miR-376c level was validated as a bio- promotes carcinogenesis and serves as a plasma marker for gastric carcinoma. PLoS ONE 12(5): marker for gastric carcinoma, including early stage tumors. The induction of miR-376c was e0177346. -

Core Circadian Clock Transcription Factor BMAL1 Regulates Mammary Epithelial Cell
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.23.432439; this version posted February 23, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Title: Core circadian clock transcription factor BMAL1 regulates mammary epithelial cell 2 growth, differentiation, and milk component synthesis. 3 Authors: Theresa Casey1ǂ, Aridany Suarez-Trujillo1, Shelby Cummings1, Katelyn Huff1, 4 Jennifer Crodian1, Ketaki Bhide2, Clare Aduwari1, Kelsey Teeple1, Avi Shamay3, Sameer J. 5 Mabjeesh4, Phillip San Miguel5, Jyothi Thimmapuram2, and Karen Plaut1 6 Affiliations: 1. Department of Animal Science, Purdue University, West Lafayette, IN, USA; 2. 7 Bioinformatics Core, Purdue University; 3. Animal Science Institute, Agriculture Research 8 Origination, The Volcani Center, Rishon Letsiyon, Israel. 4. Department of Animal Sciences, 9 The Robert H. Smith Faculty of Agriculture, Food, and Environment, The Hebrew University of 10 Jerusalem, Rehovot, Israel. 5. Genomics Core, Purdue University 11 Grant support: Binational Agricultural Research Development (BARD) Research Project US- 12 4715-14; Photoperiod effects on milk production in goats: Are they mediated by the molecular 13 clock in the mammary gland? 14 ǂAddress for correspondence. 15 Theresa M. Casey 16 BCHM Room 326 17 175 South University St. 18 West Lafayette, IN 47907 19 Email: [email protected] 20 Phone: 802-373-1319 21 22 bioRxiv preprint doi: https://doi.org/10.1101/2021.02.23.432439; this version posted February 23, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

ARID2 Deficiency Promotes Tumor Progression and Is Associated with Higher
bioRxiv preprint doi: https://doi.org/10.1101/2020.01.10.898726; this version posted January 10, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. ARID2 deficiency promotes tumor progression and is associated with higher sensitivity to PARP inhibition in lung cancer. Thaidy Moreno1, Laura González-Silva1, Beatriz Monterde1, Isabel Betancor-Fernández2, Carlos Revilla1, Antonio Agraz-Doblas1, Javier Freire3, Pablo Isidro4, Laura Quevedo1, Santiago Montes-Moreno3, Laura Cereceda3, Aurora Astudillo4, Berta Casar1,6, Piero Crespo1,6, Cristina Morales Torres5, Paola Scaffidi5,7, Javier Gomez-Roman3, Eduardo Salido2, and Ignacio Varela1* 1) Instituto de Biomedicina y Biotecnología de Cantabria. Universidad de Cantabria-CSIC. Santander, Spain. 2) Departamento de Patología, Centro de Investigación Biomédica en Red de Enfermedades Raras by impairing DNA repair. Importantly, our results indicate that ARID2-deficiency could be(CIBERER). Tenerife, Spain. 3) Servicio de Anatomía Patológica y Biobanco Valdecilla. HUMV/IDIVAL. Santander, Spain. 4) Biobanco del Principado de Asturias (BBPA). Hospital Universitario Central de Asturias. Oviedo, Spain. 5) Cancer Epigenetics Laboratory, The Francis Crick Institute. London, UK. 6) Centro de Investigación Biomédica en Red de Cáncer (CIBERONC) 7) UCL Cancer Institute, London, UK. * To whom correspondence should be addressed: Ignacio Varela Instituto de Biomedicina y Biotecnología de Cantabria Universidad de Cantabria Albert Einstein 22, 39011 Santander Spain. Ph. +34 942203931 Email. [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.01.10.898726; this version posted January 10, 2020. -

Emerging Role of SWI/SNF Complex Deficiency As a Target of Immune
Zhou et al. Oncogenesis (2021) 10:3 https://doi.org/10.1038/s41389-020-00296-6 Oncogenesis REVIEW ARTICLE Open Access Emerging role of SWI/SNF complex deficiency as a target of immune checkpoint blockade in human cancers Min Zhou 1,2,JianlongYuan1,2,YaqiDeng1,2,XianqunFan 1,2 and Jianfeng Shen 1,2 Abstract Mammalian SWI/SNF complex is a key chromatin remodeler that reshapes nucleosomes and regulates DNA accessibility. Mutations in SWI/SNF subunits are found in a broad spectrum of human cancers; however, the mechanisms of how these aberrations of SWI/SNF complex would impact tumorigenesis and cancer therapeutics remain to be elucidated. Studies have demonstrated that immune checkpoint blockade (ICB) therapy is promising in cancer treatment. Nevertheless, suitable biomarkers that reliably predict the clinical response to ICB are still lacking. Emerging evidence has suggested that SWI/SNF components play novel roles in the regulation of anti-tumor immunity, and SWI/SNF deficiency can be therapeutically targeted by ICB. These findings manifest the prominence of the SWI/SNF complex as a stratification biomarker that predicts treatment (therapeutic) response to ICB. In this review, we summarize the recent advances in ICB therapy by harnessing the cancer-specific vulnerability elicited by SWI/SNF deficiency. We provide novel insights into a comprehensive understanding of the underlying mechanisms by which SWI/SNF functions as a modulator of anti-tumor immunity. 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; Introduction mutations or defects could be exploited for therapeutic The mammalian switch/sucrose non-fermentable (SWI/ purposes6. SNF) family is a multi-subunit chromatin remodeling Cancer immunotherapy especially immune checkpoint complex that utilizes the energy of ATP hydrolysis to blockade (ICB) has recently become one of the most pro- remodel nucleosomes and regulates DNA accessibility in minent therapeutics for human cancers7. -

Mechanisms of Immune Checkpoint Inhibitor Circumstances Mechanism Circumstances Mechanism
Mechanisms of Immune Checkpoint Inhibitor Circumstances Mechanism Circumstances Mechanism PD-L1– Nonreversible and severe T-cell exhaustion Association with Loss-of-function mutations in chromatin overexpressed neoantigen remodeler genes (PBRM1, ARID2 and BRD7) tumors Coexpression of inhibitory receptors (LAG-3, overexpression sensitize tumors to ICB and increase TIM-3, TIGIT, VISTA, and BTLA) accessibility to regulatory elements of IFN-γ– by genetic inducible genes. Decreased ratio of TILs to Tregs and MDSCs alterations in mammalian Loss of ARID1A leads to increased MSI with Altered metabolism through IDO & increased SWI/SNF adenosine production inability to recruit mismatch repair genes chromatin during DNA repair, increasing mutational Mutations in PTEN, EGFR, and MYC remodeling burden and neoantigen load. complexes PD-1 and CTLA-4 Immunoediting with loss of neoantigens Stability of chromatin remodeling complexes in inhibitor tumors contributes to ICB resistance combination Deletions or mutations in JAK1/2, IFNGR1/2, & therapy IRF1 High mutation Decreased antigen presentation secondary to MHC, β2-microglobulin, and NLRC5 alterations Decreased T-cell priming and DC dysfunction overload tumors AK1/2 mutations and decreased IFN-γ signaling Aberrant WNT/β-catenin signaling High copy number loss of tumor suppressor Upregulation of alternate inhibitory genes checkpoints Mechanism of Drug Resistance ctDNA for MRD in Colon Cancer ctDNA for MRD in Colon Cancer Factors associated with TTR From Cancer Stem Cell to See the Future Research in Colorectal Cancer Colon Cancer has the Characteristics and Advantages of both Stemness of Cancer Cell and Intestinal System CSC + ISC CCSC Cancer Stem Cell • Possess special biological properties • Very Important to understand the cellular • Long-term self-renewal capacity survival mechanisms • Multi-lineage differentiation • To eradicate cancer stem cells and • Resistance to conventional preventing chemotherapy and radiotherapy chemotherapy and radiotherapy resistance. -

RESEARCH ARTICLE Analysis of ARID2 Gene Mutation in Oral
DOI:10.22034/APJCP.2017.18.10.2679 ARID2 in Oral Squamous Cell Carcinoma RESEARCH ARTICLE Analysis of ARID2 Gene Mutation in Oral Squamous Cell Carcinoma Lakshmi Prabha Das1*, Raghuram Hari Pitty1, Kannan Asokan1, Krithika C-L1, Anandi M-S1, Arvind Ramanathan2 Abstract Background: The ARID2 gene, encoding a sub unit of the chromatin remodelling complex, has a possible tumour suppressor function and has been found to be frequently mutated in various tumours, including gingivo buccal oral squamous cell carcinomas. The present study was designed to analyse the presence of ARID2 gene mutations in the distinct genetic South Indian (Dravidian) population. Materials and Methods: Genomic DNA from thirty biopsy tissue samples of histopathologically confirmed cases of oral squamous cell carcinoma (OSCC) were subjected to PCR amplification with intronic primers encompassing exons 19 and 20 of ARID2. Subsequently, the PCR amplicons were purified and subjected to Sanger sequencing using forward primers for analysis of mutational status. Results: Our study yielded a 6% occurrence of mutations in the ARID2 gene among the thirty OSCC samples. Two samples showed a C(5174)A nonsense mutation whereby the “C” nucleotide was substituted with an “A” nucleotide at position 5174, resulting in the conversion of serine amino acid at codon 1725 to a premature STOP codon. Conclusion: Identification of ARID2 gene mutations in OSCCs in this distinct ethnic population reaffirms that aberrations in the chromatin remodelling complex could indeed also contribute to tumorigenesis, thus providing new insights for future research. Keywords: Oral squamous cell carcinoma- ARID2- chromatin remodelling complex- PBAF- mutation Asian Pac J Cancer Prev, 18 (10), 2679-2681 Introduction therapies and also predict responses to various treatment modalities (Bhatt et al., 2010). -

ARID2 Chromatin Remodeler in Hepatocellular Carcinoma
cells Review ARID2 Chromatin Remodeler in Hepatocellular Carcinoma Robin Loesch 1,2, Linda Chenane 1,2 and Sabine Colnot 1,2,* 1 INSERM, Centre de Recherche des Cordeliers (CRC), Sorbonne Université, Université de Paris, F-75006 Paris, France; [email protected] (R.L.); [email protected] (L.C.) 2 Equipe labellisée “Ligue Nationale Contre le Cancer”, F-75013 Paris, France * Correspondence: [email protected] Received: 30 July 2020; Accepted: 16 September 2020; Published: 23 September 2020 Abstract: Chromatin remodelers are found highly mutated in cancer including hepatocellular carcinoma. These mutations frequently occur in ARID (AT-rich Interactive Domain) genes, encoding subunits of the ATP-dependent SWI/SNF remodelers. The increasingly prevalent complexity that surrounds the functions and specificities of the highly modular BAF (BG1/BRM-associated factors) and PBAF (polybromo-associated BAF) complexes, including ARID1A/B or ARID2, is baffling. The involvement of the SWI/SNF complexes in diverse tissues and processes, and especially in the regulation of gene expression, multiplies the specific outcomes of specific gene alterations. A better understanding of the molecular consequences of specific mutations impairing chromatin remodelers is needed. In this review, we summarize what we know about the tumor-modulating properties of ARID2 in hepatocellular carcinoma. Keywords: ARID2; hepatocellular carcinoma; cancer; chromatin; SWI/SNF 1. Introduction to the SWI/SNF Chromatin Remodeling Complexes Gene expression regulation is far from being a random phenomenon. It requires covalent modifications targeting both DNA via methylation, and histones via post-translational modifications (PTM), which shape the nucleosomes and control the DNA compaction into chromatin and the recruitment of specific transcription factors. -
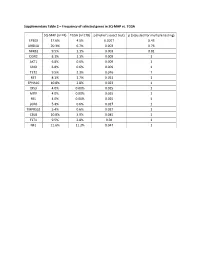
Supplementary Table 2 – Frequency of Selected Genes in SQ-MAP Vs
Supplementary Table 2 – Frequency of selected genes in SQ-MAP vs. TCGA SQ-MAP (n=74) TCGA (n=178) p (Fisher’s exact test) p (adjusted for multiple testing) EP300 17.6% 4.5% 0.0017 0.43 ARID1A 20.3% 6.7% 0.003 0.76 NFKB1 9.5% 1.1% 0.003 0.81 DDR2 8.1% 1.1% 0.009 1 AKT1 6.8% 0.6% 0.009 1 SMO 6.8% 0.6% 0.009 1 TET2 9.5% 2.3% 0.016 1 RET 8.1% 1.7% 0.021 1 EPHA10 10.8% 2.8% 0.023 1 DIS3 4.0% 0.00% 0.025 1 MITF 4.0% 0.00% 0.025 1 REL 4.0% 0.00% 0.025 1 LGR6 5.4% 0.6% 0.027 1 TMPRSS2 5.4% 0.6% 0.027 1 CBLB 10.8% 3.9% 0.045 1 FLT4 9.5% 2.8% 0.04 1 NF1 21.6% 11.2% 0.047 1 Supplementary Table 3 – Clinical characteristics of patients with resected brain metastases ID Age Sex Race Smoking status Pack-years Treatment cisplatin + docetaxel -> cisplatin + PP1 51 Female White Former 10 gemcitabine -> sequential RT; gemcitabine; nab-paclitaxel PP2 88 Male White Former 50 carboplatin + paclitaxel PP3 62 Female White Current 33 cisplatin + etoposide + concurrent RT PP4 66 Female White Former 20 cisplatin + gemcitabine carboplatin + paclitaxel + concurrent PP5 67 Female White Former 50 RT PP6 77 Male White Current 53 carboplatin + paclitaxel Supplementary Table 4 – Genes exhibiting clonal evolution from primary lung tumors and matched brain metastases. -

Genomic Landscape and Genetic Heterogeneity in Gastric Adenocarcinoma Revealed by Whole-Genome Sequencing
ARTICLE Received 1 May 2014 | Accepted 3 Oct 2014 | Published 19 Nov 2014 DOI: 10.1038/ncomms6477 Genomic landscape and genetic heterogeneity in gastric adenocarcinoma revealed by whole-genome sequencing Swee Seong Wong1,*, Kyoung-Mee Kim2,*, Jason C. Ting1,*, Kun Yu1,w, Jake Fu3, Shawn Liu4, Razvan Cristescu5, Michael Nebozhyn5, Lara Gong4, Yong Gang Yue1, Jian Wang1, Chen Ronghua5, Andrey Loboda5, James Hardwick5,w, Xiaoqiao Liu5, Hongyue Dai5, Jason Gang Jin3, Xiang S. Ye1, So Young Kang2,InGuDo2, Joon Oh Park6, Tae Sung Sohn7, Christoph Reinhard1, Jeeyun Lee6, Sung Kim7 & Amit Aggarwal1 Gastric cancer (GC) is the second most common cause of cancer-related deaths. It is known to be a heterogeneous disease with several molecular and histological subtypes. Here we perform whole-genome sequencing of 49 GCs with diffuse (N ¼ 31) and intestinal (N ¼ 18) histological subtypes and identify three mutational signatures, impacting TpT, CpG and TpCp[A/T] nucleotides. The diffuse-type GCs show significantly lower clonality and smaller numbers of somatic and structural variants compared with intestinal subtype. We further divide the diffuse subtype into one with infrequent genetic changes/low clonality and another with relatively higher clonality and mutations impacting TpT dinucleotide. Notably, we dis- cover frequent and exclusive mutations in Ephrins and SLIT/ROBO signalling pathway genes. Overall, this study delivers new insights into the mutational heterogeneity underlying distinct histologic subtypes of GC that could have important implications for future research in the diagnosis and treatment of GC. 1 Lilly Research Labs, Eli Lilly and Co, Indianapolis, Indiana 46285, USA. 2 Department of Pathology & Translational Genomics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul 135-710, South Korea. -

Interplay Between Cofactors and Transcription Factors in Hematopoiesis and Hematological Malignancies
Signal Transduction and Targeted Therapy www.nature.com/sigtrans REVIEW ARTICLE OPEN Interplay between cofactors and transcription factors in hematopoiesis and hematological malignancies Zi Wang 1,2, Pan Wang2, Yanan Li2, Hongling Peng1, Yu Zhu2, Narla Mohandas3 and Jing Liu2 Hematopoiesis requires finely tuned regulation of gene expression at each stage of development. The regulation of gene transcription involves not only individual transcription factors (TFs) but also transcription complexes (TCs) composed of transcription factor(s) and multisubunit cofactors. In their normal compositions, TCs orchestrate lineage-specific patterns of gene expression and ensure the production of the correct proportions of individual cell lineages during hematopoiesis. The integration of posttranslational and conformational modifications in the chromatin landscape, nucleosomes, histones and interacting components via the cofactor–TF interplay is critical to optimal TF activity. Mutations or translocations of cofactor genes are expected to alter cofactor–TF interactions, which may be causative for the pathogenesis of various hematologic disorders. Blocking TF oncogenic activity in hematologic disorders through targeting cofactors in aberrant complexes has been an exciting therapeutic strategy. In this review, we summarize the current knowledge regarding the models and functions of cofactor–TF interplay in physiological hematopoiesis and highlight their implications in the etiology of hematological malignancies. This review presents a deep insight into the physiological and pathological implications of transcription machinery in the blood system. Signal Transduction and Targeted Therapy (2021) ;6:24 https://doi.org/10.1038/s41392-020-00422-1 1234567890();,: INTRODUCTION by their ATPase subunits into four major families, including the Hematopoiesisisacomplexhierarchicaldifferentiationprocessthat SWI/SNF, ISWI, Mi-2/NuRD, and INO80/SWR1 families. -

(NGS) for Primary Endocrine Resistance in Breast Cancer Patients
Int J Clin Exp Pathol 2018;11(11):5450-5458 www.ijcep.com /ISSN:1936-2625/IJCEP0084102 Original Article Impact of next-generation sequencing (NGS) for primary endocrine resistance in breast cancer patients Ruoyang Li1*, Tiantian Tang1*, Tianli Hui1, Zhenchuan Song1, Fugen Li2, Jingyu Li2, Jiajia Xu2 1Breast Center, Fourth Hospital of Hebei Medical University, Shijiazhuang, China; 2Institute of Precision Medicine, 3D Medicines Inc., Shanghai, China. *Equal contributors. Received August 15, 2018; Accepted September 22, 2018; Epub November 1, 2018; Published November 15, 2018 Abstract: Multiple mechanisms have been detected to account for the acquired resistance to endocrine therapies in breast cancer. In this study we retrospectively studied the mechanism of primary endocrine resistance in estrogen receptor positive (ER+) breast cancer patients by next-generation sequencing (NGS). Tumor specimens and matched blood samples were obtained from 24 ER+ breast cancer patients. Fifteen of them displayed endocrine resistance, including recurrence and/or metastases within 24 months from the beginning of endocrine therapy, and 9 pa- tients remained sensitive to endocrine therapy for more than 5 years. Genomic DNA of tumor tissue was extracted from formalin-fixed paraffin-embedded (FFPE) tumor tissue blocks. Genomic DNA of normal tissue was extracted from peripheral blood mononuclear cells (PBMC). Sequencing libraries for each sample were prepared, followed by target capturing for 372 genes that are frequently rearranged in cancers. Massive parallel -

Expression and Prognostic Values of ARID Family Members in Breast Cancer
www.aging-us.com AGING 2021, Vol. 13, No. 4 Research Paper Expression and prognostic values of ARID family members in breast cancer Jinfeng Zhang1,*, Siyu Hou1,*, Zilong You1, Guozheng Li1, Shouping Xu1, Xianyong Li3, Xianyu Zhang1, Bo Lei1, Da Pang1,2 1Department of Breast Surgery, Harbin Medical University Cancer Hospital, Harbin, China 2Heilongjiang Academy of Medical Sciences, Harbin, China 3Department of Critical Care Medicine, The First Affiliated Hospital of Harbin Medical University, Harbin, China *Equal contribution Correspondence to: Da Pang, Bo Lei; email: [email protected]; [email protected], https://orcid.org/0000-0002- 5613-5233 Keywords: biomarkers, tumor, breast neoplasms, prognosis Received: April 20, 2020 Accepted: December 3, 2020 Published: February 11, 2021 Copyright: © 2021 Zhang et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT The ARID family is a superfamily of 15 members containing a domain that interacts with AT-rich DNA elements. However, the expression and prognostic roles of each ARID in breast cancer are still elusive. We used the TCGA and Kaplan-Meier plotter databases to assess the expression and prognostic values of ARID mRNA levels in breast cancer respectively. In the present study, 6 members were significantly lower in tumor tissues than those in the normal tissues, while 6 members were significantly higher. Further assessment of ARID expression in breast cancer with different molecular subtypes, 3 members were significantly higher in no- luminal molecular subtype than those in the luminal molecular subtype, and 6 members were significantly higher.