Effects of Halogen, Chalcogen, Pnicogen, and Tetrel Bonds on IR and NMR Spectra
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Alternate Graphical Representation of Periodic Table of Chemical Elements Mohd Abubakr1, Microsoft India (R&D) Pvt
An Alternate Graphical Representation of Periodic table of Chemical Elements Mohd Abubakr1, Microsoft India (R&D) Pvt. Ltd, Hyderabad, India. [email protected] Abstract Periodic table of chemical elements symbolizes an elegant graphical representation of symmetry at atomic level and provides an overview on arrangement of electrons. It started merely as tabular representation of chemical elements, later got strengthened with quantum mechanical description of atomic structure and recent studies have revealed that periodic table can be formulated using SO(4,2) SU(2) group. IUPAC, the governing body in Chemistry, doesn‟t approve any periodic table as a standard periodic table. The only specific recommendation provided by IUPAC is that the periodic table should follow the 1 to 18 group numbering. In this technical paper, we describe a new graphical representation of periodic table, referred as „Circular form of Periodic table‟. The advantages of circular form of periodic table over other representations are discussed along with a brief discussion on history of periodic tables. 1. Introduction The profoundness of inherent symmetry in nature can be seen at different depths of atomic scales. Periodic table symbolizes one such elegant symmetry existing within the atomic structure of chemical elements. This so called „symmetry‟ within the atomic structures has been widely studied from different prospects and over the last hundreds years more than 700 different graphical representations of Periodic tables have emerged [1]. Each graphical representation of chemical elements attempted to portray certain symmetries in form of columns, rows, spirals, dimensions etc. Out of all the graphical representations, the rectangular form of periodic table (also referred as Long form of periodic table or Modern periodic table) has gained wide acceptance. -

25 WORDS CHLORINE Chlorine, Cl, Is a Very Poisonous Green Gas That's
25 WORDS CHLORINE Chlorine, Cl, is a very poisonous green gas that's extremely reactive. It's used for sanitizing, purifying, and was used as a weapon during World War I by the Germans. But in chemistry, it is an oxidizer. Chlorine, Cl, is a green gaseous element with an atomic number of 17. This halogen is a powerful oxidant and used to produce many things, such as cleaning products. Chlorine; it's chemical symbol is Cl. Chloride is abundant in nature and necessary for life but a large amount can cause choking and and poisoning. It's mainly used for water purification but has other uses. Chlorine is a halogen and to test if it has a halogen, we use the Beilstein Copper Wire Test. It is also used to produce safe drinking water. Chlorine, atomic number seventeen, is a halogen that is found in table salt, NaCl, making it essential to life. However, pure chlorine, Cl2, is a poisonous gas, detectable at even 1 ppm. Chlorine, (Symbol Cl), belongs to the halogen family of elements, found in group 17 on the periodic table. Chlorine has an atomic number of 17 and atomic weight of 35.453. Chlorine is the 17th element on the periodic table, and is in the "Halogens" group, which has a tendency to gain one electron to form anions. Its anion can be found commonly in table salt Chlorine (symbolized Cl) is the chemical element with atomic number 17. Clorine is a powerful oxidant and is used in bleaching and disinfectants. It is a pale yellow-green gas that has a specific strong smell. -

Driving Halogen Lamps Application Note
Application Note 1604 Driving Halogen Lamps Abstract: This application note looks at the suitability of the Ultimod wide trim powerMods for applications driving Halogen lamps. An incandescent lamp generates light by heating is that the powerMod will go into a protective a tungsten wire or filament until it glows (at current limit. The characteristics of this current around 2,500 ºC) by passing an electric current limit are shown in Figure 1 below. through it. A halogen lamp is basically a modified version of an incandescent lamp. The difference is that the bulb of a halogen lamp has a small amount of a halogen gas added. The presence of this halogen in the bulb produces a chemical reaction (known as the halogen cycle) that redeposists tungsten evaporated by heating back onto the filament. In a standard incandescent the constant evaporation leads to the eventual failure of the lamp as the filament progressively thins and breaks or “burns out”. Since in a halogen lamp the tungsten is redeposited back on the filament Figure 1 Current Limit Characteristics its lifetime is extended, and it also be heated to a higher temperature (in the region of 3,000 ºC), which increases its efficiency. You can see from Figure 1 that when we increase the load (by reducing the loads The high operational temperature of the filament resistance) and the current increases above the results in a challenge for a constant voltage set current limit of the modules we enter a mode power supplies like the Ultimod due to the of operation known as straight line current different resistance of the tungsten filament limiting where the current is held constant and at room temperature and its resistance at 3,000 the voltage is reduced. -

Polymorphism, Halogen Bonding, and Chalcogen Bonding in the Diiodine Adducts of 1,3- and 1,4-Dithiane
molecules Article Polymorphism, Halogen Bonding, and Chalcogen Bonding in the Diiodine Adducts of 1,3- and 1,4-Dithiane Andrew J. Peloquin 1, Srikar Alapati 2, Colin D. McMillen 1, Timothy W. Hanks 2 and William T. Pennington 1,* 1 Department of Chemistry, Clemson University, Clemson, SC 29634, USA; [email protected] (A.J.P.); [email protected] (C.D.M.) 2 Department of Chemistry, Furman University, Greenville, SC 29613, USA; [email protected] (S.A.); [email protected] (T.W.H.) * Correspondence: [email protected] Abstract: Through variations in reaction solvent and stoichiometry, a series of S-diiodine adducts of 1,3- and 1,4-dithiane were isolated by direct reaction of the dithianes with molecular diiodine in solution. In the case of 1,3-dithiane, variations in reaction solvent yielded both the equatorial and the axial isomers of S-diiodo-1,3-dithiane, and their solution thermodynamics were further studied via DFT. Additionally, S,S’-bis(diiodo)-1,3-dithiane was also isolated. The 1:1 cocrystal, (1,4-dithiane)·(I2) was further isolated, as well as a new polymorph of S,S’-bis(diiodo)-1,4-dithiane. Each structure showed significant S···I halogen and chalcogen bonding interactions. Further, the product of the diiodine-promoted oxidative addition of acetone to 1,4-dithiane, as well as two new cocrystals of 1,4-dithiane-1,4-dioxide involving hydronium, bromide, and tribromide ions, was isolated. Keywords: crystal engineering; chalcogen bonding; halogen bonding; polymorphism; X-ray diffraction Citation: Peloquin, A.J.; Alapati, S.; McMillen, C.D.; Hanks, T.W.; Pennington, W.T. -

Of the Periodic Table
of the Periodic Table teacher notes Give your students a visual introduction to the families of the periodic table! This product includes eight mini- posters, one for each of the element families on the main group of the periodic table: Alkali Metals, Alkaline Earth Metals, Boron/Aluminum Group (Icosagens), Carbon Group (Crystallogens), Nitrogen Group (Pnictogens), Oxygen Group (Chalcogens), Halogens, and Noble Gases. The mini-posters give overview information about the family as well as a visual of where on the periodic table the family is located and a diagram of an atom of that family highlighting the number of valence electrons. Also included is the student packet, which is broken into the eight families and asks for specific information that students will find on the mini-posters. The students are also directed to color each family with a specific color on the blank graphic organizer at the end of their packet and they go to the fantastic interactive table at www.periodictable.com to learn even more about the elements in each family. Furthermore, there is a section for students to conduct their own research on the element of hydrogen, which does not belong to a family. When I use this activity, I print two of each mini-poster in color (pages 8 through 15 of this file), laminate them, and lay them on a big table. I have students work in partners to read about each family, one at a time, and complete that section of the student packet (pages 16 through 21 of this file). When they finish, they bring the mini-poster back to the table for another group to use. -

Halides and Halogens. What Do I Need to Know? John Vivari, Nordson EFD
Halides and Halogens. What do I need to know? John Vivari, Nordson EFD Abstract With halogen-containing substances in the public eye due to scrutiny by the European Union and a variety of non- governmental organizations (NGOs) as possible additions to the list of substances banned from electronics, we at EFD have received numerous inquiries from customers asking how this subject will affect them and their processes. Having just overcome the hurdle of RoHS (Restriction of Hazardous Substances), they want to know what halogens and halides are, and what changes they should be prepared for if required to stop using them. Halide-free materials are not new. Some segments of the electronics industry have been sensitive to halides and their significance for decades. This paper will give the reader a working knowledge of halogens and halides. Armed with this education, the reader will be able to make informed decisions when required to use halogen-free materials, either because regulations dictate it or social pressure makes acceptance preferable to resistance. Key Words: halide, halogen, bromine, chlorine, flame retardant, RoHS What are halogens and halides? damage. Brominated flame retardant use is not limited to electronics. It is also in common usage in furniture, At their most basic level, halogens are the electronegative construction materials and textiles. elements in column 17 of the periodic table, including fluorine (F), chlorine, (Cl), bromine (Br), iodine (I) and Other sources of halogens in circuit boards include astatine (At). In electronics fiberglass sizing, epoxy curing agents and accelerators, applications, iodine and resin wetting and de-foaming agents, flux residues, and astatine are rarely if ever contamination from handling. -

The Halogens Family Word List
The Halogens Family Word list Halogen – halogenas Any of the electronegative elements, fluorine, chlorine, iodine, bromine, and astatine, that form binary salts by direct union with metals. Fluorine – floras The most reactive nonmetallic element, a pale-yellow, corrosive, toxic gas that occurs combined, especially in fluorite, cryolite, phosphate rock, and other minerals. Chlorine – chloras A halogen element, a heavy, greenish-yellow, incombustible, water-soluble, poisonous gas. Bromine – bromas An element that is a dark-reddish, fuming, toxic liquid and a member of the halogen family. Iodine – jodas A nonmetallic halogen element occurring at ordinary temperatures as a grayish-black crystalline solid. Astatine – astatas A highly unstable radioactive element, the heaviest of the halogen series, that resembles iodine in solution. Bleach - baliklis A chemical agent used for bleaching. Ion – jonas An atom or a group of atoms that has acquired a net electric charge by gaining or losing one or more electrons. Compound – junginys A pure, macroscopically homogeneous substance consisting of atoms or ions of two or more different elements in definite proportions that cannot be separated by physical means. Solid – kieta medžiaga Having three dimensions (length, breadth, and thickness), as a geometrical body or figure. Vapour – garai Particles of moisture or other substance suspended in air and visible as clouds, smoke, etc. Conductor – laidininkas A substance, body, or device that readily conducts heat, electricity, sound, etc. Hydrogen – vandenilis A colorless, odorless, flammable gas that combines chemically with oxygen to form water. Sodium – natris A soft, silver-white, metallic element that oxidizes rapidly in moist air, occurring in nature only in the combined state. -

Pre and Post Tests Use the Periodic Table to Answer These Questions
Pre and Post Tests Use the periodic table to answer these questions. MULTIPLE CHOICE 1. Neon is an example of a(n): A. Alkali Metal B. Noble Gas C. Halogen D. Rare Earth Metal 2. An element that has two valence or outer level electrons would be considered a(n): A. Nonmetal B. Noble Gas C. Halogen D. Metal 3. Sodium is in the same group or family as: A. Lithium B. Helium C. Calcium D. Thorium 4. Iodine is an example of a(n): A. Noble Gas B. Halogen C. Alkali Metal D. Rare Earth Metal 5. If an element is a gas at room temperature, then it must be a(n): A. Alkali Metal B. Nonmetal C. Halogen D. Alkaline Earth Metal 6. If an element has one to three valence or outer level electrons, then it is a(n): A. Nonmetal B. Halogen C. Noble Gas D. Metal 7. Which of the following elements belongs to the group that includes the most active metals? A. Aluminum B. Sodium C. Iron D. Mercury GRIDDED RESPONSE 8. How many valence or outer level electrons does an atom of the element argon have? _____________ SHORT RESPONSE 9. Barium and magnesium are both found in the same group in the periodic table. Name two properties that they would be expected to share. EXTENDED RESPONSE 10. Chlorine and bromine are both found in the same group in the periodic table. Name this group. Are these elements metals or nonmetals? How many valence electrons do they have? Name a property that these two elements would be expected to share. -
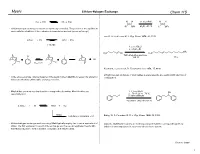
Lithium Halogen Exchange #1 Revised
Myers Lithium-Halogen Exchange Chem 115 RLi + R'X RX + R'Li H H 1.1 eq n-BuLi H H Br OEt Et2O, !80 °C Li OEt Lithium-halogen exchange reactions are kinetically controlled. The position of the equilibrium varies with the stabilities of the carbanion intermediates involved (sp >> sp2 >> sp3) Lau, K. S.; Schlosser, M. J. Org. Chem. 1978, 43, 1595. n-PrLi + PhI n-PrI + PhLi 1:10,000 1. 2 eq t-BuLi 2. n-C8H17Br CH3 H3C Br H3C Li I THF-ethyl ether-pentane !120 °C 77% LiI I Li Keq << 1 I I Neumann, H.; Seebach, D. Tetrahedron Lett. 1976, 17, 4839. Lithium-halogen exchange of vinyl halides is stereospecific, proceeding with retention of In the above example, internal trapping of the newly formed alkyllithium reagent by alkylation configuration. drives an otherwise unfavorable exchange reaction. Alkyliodides are more reactive than the corresponding bromides. Alkylchlorides are 1. 2.1 eq t-BuLi OH essentially inert. 2. !78 " 23 " !78 °C I 3. benzaldehyde n-pentane-ethyl ether (3:2) 2 t-BuLi + RI t-BuI + RLi t-BuLi isobutene + isobutane + LiI Bailey, W. F.; Punzalan, E. R. J. Org. Chem. 1990, 55, 5404. Lithium-halogen exchange reactions using t-BuLi typically employ two or more equivalents of Aliphatic alkyllithium reagents are normally prepared from the corresponding primary t-BuLi. The first equivalent is used for the exchange and the second equivalent reacts with iodides at low temperature in a pentane-ether solvent system. the t-BuI produced, to form isobutene, isobutane, and lithium iodide. -

3 Families of Elements
Name Class Date CHAPTER 5 The Periodic Table SECTION 3 Families of Elements KEY IDEAS As you read this section, keep these questions in mind: • What makes up a family of elements? • What properties do the elements in a group share? • Why does carbon form so many compounds? What Are Element Families? Recall that all elements can be classified into three READING TOOLBOX categories: metals, nonmetals, and semiconductors. Organize As you read Scientists classify the elements further into five families. this section, create a chart The atoms of all elements in most families have the same comparing the different number of valence electrons. Thus, members of a family families of elements. Include examples of each family in the periodic table share some properties. and describe the common properties of elements in the Group number Number of valence Name of family family. electrons Group 1 1 Alkali metals Group 2 2 Alkaline-earth metals READING CHECK Groups 3–12 varied Transition metals 1. Identify In general, what Group 17 7 Halogens do all elements in the same family have in common? Group 18 8 (except helium, Noble gases which has 2) What Are the Families of Metals? Many elements are classified as metals. Recall that metals can conduct heat and electricity. Most metals can be stretched and shaped into flat sheets or pulled into wires. Families of metals include the alkali metals, the alkaline-earth metals, and the transition metals. THE ALKALI METALS READING CHECK The elements in Group 1 form a family called the 2. Explain Why are alkali alkali metals. -

Halogen-Metal Exchange Reactions of Bromoaryl-Substituted Β-Lactams
TCNJ JOURNAL OF STUDENT SCHOLARSHIP VOLUME IX APRIL, 2007 HALOGEN-METAL EXCHANGE REACTIONS OF BROMOARYL- SUBSTITUTED β-LACTAMS Author: Maryll Geherty Faculty Sponsor: David A. Hunt, Department of Chemistry ABSTRACT Compounds possessing the β-lactam ring system have long been of intense interest because of their antibiotic properties.1,2 Among the more well-known examples of compounds belonging to this class are penicillins and cephalosporins. This project focused on determining the feasibility of elaborating bromoaryl-substituted β-lactam systems via a halogen-metal exchange reaction in order to make novel derivatives which would be difficult to synthesize otherwise because of to the highly electrophilic nature of the β-lactam ring system. INTRODUCTION/STATEMENT OF THE PROBLEM In the 1970s, W. E. Parham and his research group demonstrated that halogen-metal exchange reactions were possible on aryl bromides to form aryl compounds bearing electrophilic groups (Scheme 1).3 These reactions were conducted at low temperatures of -100 ° C using either n- butyllithium or t-butyllithium as the exchange reagent. These studies established a new reaction paradigm: halogen-lithium exchange reactions can occur at low temperatures chemoselectively without the formation of unwanted side products resulting from reaction of the electrophilic groups contained in the molecule with the alkylithium reagents. These side reactions would be the primary reaction pathway at higher temperatures. Scheme 1 (CH2)nX (CH ) X 2 n (CH2)nX n-C4H9Li E+ THF/-100 oC warm to 25 oC Br Li E n = 0-4 X = Br, Cl, CN, CO2H, CO2R This research project focused on determining the feasibility of conducting halogen-metal exchange reactions on bromoaryl β-lactams. -

A Brief History of the Development of the Periodic Table
This outline was created by Western Oregon University A BRIEF HISTORY OF THE DEVELOPMENT OF THE PERIODIC TABLE Although Dmitri Mendeleev is often considered the "father" of the periodic table, the work of many scientists contributed to its present form. In the Beginning A necessary prerequisite to the construction of the periodic table was the discovery of the individual elements. Although elements such as gold, silver, tin, copper, lead and mercury have been known since antiquity, the first scientific discovery of an element occurred in 1649 when Hennig Brand discovered phosphorous. During the next 200 years, a vast body of knowledge concerning the properties of elements and their compounds was acquired by chemists (view a 1790 article on the elements). By 1869, a total of 63 elements had been discovered. As the number of known elements grew, scientists began to recognize patterns in properties and began to develop classification schemes. Law of Triads In 1817 Johann Dobereiner noticed that the atomic weight of strontium fell midway between the weights of calcium and barium, elements possessing similar chemical properties. In 1829, after discovering the halogen triad composed of chlorine, bromine, and iodine and the alkali metal triad of lithium, sodium and potassium he proposed that nature contained triads of elements the middle element had properties that were an average of the other two members when ordered by the atomic weight (the Law of Triads). This new idea of triads became a popular area of study. Between 1829 and 1858 a number of scientists (Jean Baptiste Dumas, Leopold Gmelin, Ernst Lenssen, Max von Pettenkofer, and J.P.