Kinase Domain Activation Through Gene Rearrangement in Multiple Myeloma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Significant Shortest Paths for the Detection of Putative Disease Modules
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.01.019844; this version posted April 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. SIGNIFICANT SHORTEST PATHS FOR THE DETECTION OF PUTATIVE DISEASE MODULES Daniele Pepe1 1Department of Oncology, KU Leuven, LKI–Leuven Cancer Institute, Leuven, Belgium Email address: DP: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.04.01.019844; this version posted April 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Keywords Structural equation modeling, significant shortest paths, pathway analysis, disease modules. Abstract Background The characterization of diseases in terms of perturbated gene modules was recently introduced for the analysis of gene expression data. Some approaches were proposed in literature, but many times they are inductive approaches. This means that starting directly from data, they try to infer key gene networks potentially associated to the biological phenomenon studied. However they ignore the biological information already available to characterize the gene modules. Here we propose the detection of perturbed gene modules using the combination of data driven and hypothesis-driven approaches relying on biological metabolic pathways and significant shortest paths tested by structural equation modeling. -

The Impact of Chromosomal Translocation Locus and Fusion Oncogene Coding Sequence in Synovial Sarcomagenesis
Oncogene (2016) 35, 5021–5032 © 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved 0950-9232/16 www.nature.com/onc ORIGINAL ARTICLE The impact of chromosomal translocation locus and fusion oncogene coding sequence in synovial sarcomagenesis KB Jones1,2,3, JJ Barrott1,2,3, M Xie4, M Haldar5, H Jin1,2,3, J-F Zhu1,2,3, MJ Monument1,3, TL Mosbruger3,6, EM Langer5, RL Randall1,3, RK Wilson4,7,8,9, BR Cairns2,3,10, L Ding4,7,8,9 and MR Capecchi5 Synovial sarcomas are aggressive soft-tissue malignancies that express chromosomal translocation-generated fusion genes, SS18-SSX1 or SS18-SSX2 in most cases. Here, we report a mouse sarcoma model expressing SS18-SSX1, complementing our prior model expressing SS18-SSX2. Exome sequencing identified no recurrent secondary mutations in tumors of either genotype. Most of the few mutations identified in single tumors were present in genes that were minimally or not expressed in any of the tumors. Chromosome 6, either entirely or around the fusion gene expression locus, demonstrated a copy number gain in a majority of tumors of both genotypes. Thus, by fusion oncogene coding sequence alone, SS18-SSX1 and SS18-SSX2 can each drive comparable synovial sarcomagenesis, independent from other genetic drivers. SS18-SSX1 and SS18-SSX2 tumor transcriptomes demonstrated very few consistent differences overall. In direct tumorigenesis comparisons, SS18-SSX2 was slightly more sarcomagenic than SS18-SSX1, but equivalent in its generation of biphasic histologic features. Meta-analysis of human synovial sarcoma patient series identified two tumor–gentoype–phenotype correlations that were not modeled by the mice, namely a scarcity of male hosts and biphasic histologic features among SS18-SSX2 tumors. -

Interactions Between the Parasite Philasterides Dicentrarchi and the Immune System of the Turbot Scophthalmus Maximus.A Transcriptomic Analysis
biology Article Interactions between the Parasite Philasterides dicentrarchi and the Immune System of the Turbot Scophthalmus maximus.A Transcriptomic Analysis Alejandra Valle 1 , José Manuel Leiro 2 , Patricia Pereiro 3 , Antonio Figueras 3 , Beatriz Novoa 3, Ron P. H. Dirks 4 and Jesús Lamas 1,* 1 Department of Fundamental Biology, Institute of Aquaculture, Campus Vida, University of Santiago de Compostela, 15782 Santiago de Compostela, Spain; [email protected] 2 Department of Microbiology and Parasitology, Laboratory of Parasitology, Institute of Research on Chemical and Biological Analysis, Campus Vida, University of Santiago de Compostela, 15782 Santiago de Compostela, Spain; [email protected] 3 Institute of Marine Research, Consejo Superior de Investigaciones Científicas-CSIC, 36208 Vigo, Spain; [email protected] (P.P.); antoniofi[email protected] (A.F.); [email protected] (B.N.) 4 Future Genomics Technologies, Leiden BioScience Park, 2333 BE Leiden, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +34-88-181-6951; Fax: +34-88-159-6904 Received: 4 September 2020; Accepted: 14 October 2020; Published: 15 October 2020 Simple Summary: Philasterides dicentrarchi is a free-living ciliate that causes high mortality in marine cultured fish, particularly flatfish, and in fish kept in aquaria. At present, there is still no clear picture of what makes this ciliate a fish pathogen and what makes fish resistant to this ciliate. In the present study, we used transcriptomic techniques to evaluate the interactions between P. dicentrarchi and turbot leucocytes during the early stages of infection. The findings enabled us to identify some parasite genes/proteins that may be involved in virulence and host resistance, some of which may be good candidates for inclusion in fish vaccines. -

Application of a MYC Degradation
SCIENCE SIGNALING | RESEARCH ARTICLE CANCER Copyright © 2019 The Authors, some rights reserved; Application of a MYC degradation screen identifies exclusive licensee American Association sensitivity to CDK9 inhibitors in KRAS-mutant for the Advancement of Science. No claim pancreatic cancer to original U.S. Devon R. Blake1, Angelina V. Vaseva2, Richard G. Hodge2, McKenzie P. Kline3, Thomas S. K. Gilbert1,4, Government Works Vikas Tyagi5, Daowei Huang5, Gabrielle C. Whiten5, Jacob E. Larson5, Xiaodong Wang2,5, Kenneth H. Pearce5, Laura E. Herring1,4, Lee M. Graves1,2,4, Stephen V. Frye2,5, Michael J. Emanuele1,2, Adrienne D. Cox1,2,6, Channing J. Der1,2* Stabilization of the MYC oncoprotein by KRAS signaling critically promotes the growth of pancreatic ductal adeno- carcinoma (PDAC). Thus, understanding how MYC protein stability is regulated may lead to effective therapies. Here, we used a previously developed, flow cytometry–based assay that screened a library of >800 protein kinase inhibitors and identified compounds that promoted either the stability or degradation of MYC in a KRAS-mutant PDAC cell line. We validated compounds that stabilized or destabilized MYC and then focused on one compound, Downloaded from UNC10112785, that induced the substantial loss of MYC protein in both two-dimensional (2D) and 3D cell cultures. We determined that this compound is a potent CDK9 inhibitor with a previously uncharacterized scaffold, caused MYC loss through both transcriptional and posttranslational mechanisms, and suppresses PDAC anchorage- dependent and anchorage-independent growth. We discovered that CDK9 enhanced MYC protein stability 62 through a previously unknown, KRAS-independent mechanism involving direct phosphorylation of MYC at Ser . -

Comparative Analysis of Protein Expression Concomitant with DNA Methyltransferase 3A Depletion in a Melanoma Cell Line
American Journal of Analytical Chemistry, 2011, 2, 539-572 doi:10.4236/ajac.2011.25064 Published Online September 2011 (http://www.SciRP.org/journal/ajac) Comparative Analysis of Protein Expression Concomitant with DNA Methyltransferase 3A Depletion in a Melanoma Cell Line Shengnan Tang1,#, Xiaoyan Liu1,#, Tonghua Li1*, Haoyue Wang2, Jiangming Sun1, Qian Qiao1, Jun Yao3, Jian Fei2 1Department of Chemistry, Tongji University, Shanghai, China 2School of Life Science & Technology, Tongji University, Shanghai, China 3School of Medicine, Fudan University, Shanghai, China E-mail: *[email protected] Received March 17, 2011; revised May 3, 2011; accepted June 1, 2011 Abstract DNA methyltransferase 3A (Dnmt3a), a de novo methyltransferase, has attracted a great deal of attention for its important role played in tumorigenesis. We have previously demonstrated that melanoma is unable to grow in-vivo in conditions of Dnmt3a depletion in a mouse model. In this study, we cultured the Dnmt3a depletion B16 melanoma (Dnmt3a-D) cell line to conduct a comparative analysis of protein expression con-comitant with Dnmt3a depletion in a melanoma cell line. After two-dimensional separation, by gel elec- tro-phoresis and liquid chromatography, combined with mass spectrometry analysis (1DE-LC-MS/MS), the re-sults demonstrated that 467 proteins were up-regulated and 535 proteins were down-regulated in the Dnmt3a-D cell line compared to the negative control (NC) cell line. The Genome Ontology (GO) and KEGG pathway were used to further analyze the altered proteins. KEGG pathway analysis indicated that the MAPK signaling pathway exhibited a greater alteration in proteins, an interesting finding due to the close rela- tion-ship with tumorigenesis. -

Synovial Sarcoma: Recent Discoveries As a Roadmap to New Avenues for Therapy
Published OnlineFirst January 22, 2015; DOI: 10.1158/2159-8290.CD-14-1246 REVIEW Synovial Sarcoma: Recent Discoveries as a Roadmap to New Avenues for Therapy Torsten O. Nielsen 1 , Neal M. Poulin 1 , and Marc Ladanyi 2 ABSTRACT Oncogenesis in synovial sarcoma is driven by the chromosomal translocation t(X,18; p11,q11), which generates an in-frame fusion of the SWI/SNF subunit SS18 to the C-terminal repression domains of SSX1 or SSX2. Proteomic studies have identifi ed an integral role of SS18–SSX in the SWI/SNF complex, and provide new evidence for mistargeting of polycomb repression in synovial sarcoma. Two recent in vivo studies are highlighted, providing additional support for the importance of WNT signaling in synovial sarcoma: One used a conditional mouse model in which knock- out of β-catenin prevents tumor formation, and the other used a small-molecule inhibitor of β-catenin in xenograft models. Signifi cance: Synovial sarcoma appears to arise from still poorly characterized immature mesenchymal progenitor cells through the action of its primary oncogenic driver, the SS18–SSX fusion gene, which encodes a multifaceted disruptor of epigenetic control. The effects of SS18–SSX on polycomb-mediated gene repression and SWI/SNF chromatin remodeling have recently come into focus and may offer new insights into the basic function of these processes. A central role for deregulation of WNT–β-catenin sig- naling in synovial sarcoma has also been strengthened by recent in vivo studies. These new insights into the the biology of synovial sarcoma are guiding novel preclinical and clinical studies in this aggressive cancer. -

Modulation of NF-Κb Signalling by Microbial Pathogens
REVIEWS Modulation of NF‑κB signalling by microbial pathogens Masmudur M. Rahman and Grant McFadden Abstract | The nuclear factor-κB (NF‑κB) family of transcription factors plays a central part in the host response to infection by microbial pathogens, by orchestrating the innate and acquired host immune responses. The NF‑κB proteins are activated by diverse signalling pathways that originate from many different cellular receptors and sensors. Many successful pathogens have acquired sophisticated mechanisms to regulate the NF‑κB signalling pathways by deploying subversive proteins or hijacking the host signalling molecules. Here, we describe the mechanisms by which viruses and bacteria micromanage the host NF‑κB signalling circuitry to favour the continued survival of the pathogen. The nuclear factor-κB (NF-κB) family of transcription Signalling targets upstream of NF‑κB factors regulates the expression of hundreds of genes that NF-κB proteins are tightly regulated in both the cyto- are associated with diverse cellular processes, such as pro- plasm and the nucleus6. Under normal physiological liferation, differentiation and death, as well as innate and conditions, NF‑κB complexes remain inactive in the adaptive immune responses. The mammalian NF‑κB cytoplasm through a direct interaction with proteins proteins are members of the Rel domain-containing pro- of the inhibitor of NF-κB (IκB) family, including IκBα, tein family: RELA (also known as p65), RELB, c‑REL, IκBβ and IκBε (also known as NF-κBIα, NF-κBIβ and the NF-κB p105 subunit (also known as NF‑κB1; which NF-κBIε, respectively); IκB proteins mask the nuclear is cleaved into the p50 subunit) and the NF-κB p100 localization domains in the NF‑κB complex, thus subunit (also known as NF‑κB2; which is cleaved into retaining the transcription complex in the cytoplasm. -

A Novel FISH Assay for SS18–SSX Fusion Type in Synovial Sarcoma
Laboratory Investigation (2004) 84, 1185–1192 & 2004 USCAP, Inc All rights reserved 0023-6837/04 $30.00 www.laboratoryinvestigation.org A novel FISH assay for SS18–SSX fusion type in synovial sarcoma Cecilia Surace1,2, Ioannis Panagopoulos1, Eva Pa˚lsson1, Mariano Rocchi2, Nils Mandahl1 and Fredrik Mertens1 1Department of Clinical Genetics, Lund University Hospital, Lund, Sweden and 2DAPEG, Section of Genetics, University of Bari, Bari, Italy Synovial sarcoma is a morphologically, clinically and genetically distinct entity that accounts for 5–10% of all soft tissue sarcomas. The t(X;18)(p11.2;q11.2) is the cytogenetic hallmark of synovial sarcoma and is present in more than 90% of the cases. It produces three types of fusion gene formed in part by SS18 from chromosome 18 and by SSX1, SSX2 or, rarely, SSX4 from the X chromosome. The SS18–SSX fusions do not seem to occur in other tumor types, and it has been shown that in synovial sarcoma a clear correlation exists between the type of fusion gene and histologic subtype and, more importantly, clinical outcome. Previous analyses regarding the type of fusion genes have been based on PCR amplification of the fusion transcript, requiring access to good- quality RNA. In order to obtain an alternative tool to diagnose and follow this malignancy, we developed a fluorescence in situ hybridization (FISH) assay that could distinguish between the two most common fusion genes, that is, SS18–SSX1 and SS18–SSX2. The specificity of the selected bacterial artificial chromosome clones used in the detection of these fusion genes, as well as the sensitivity of the analysis in metaphase and interphase cells, was examined in a series of 28 synovial sarcoma samples with known fusion gene status. -

Characterization of the Small Molecule Kinase Inhibitor SU11248 (Sunitinib/ SUTENT in Vitro and in Vivo
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Genetik Characterization of the Small Molecule Kinase Inhibitor SU11248 (Sunitinib/ SUTENT in vitro and in vivo - Towards Response Prediction in Cancer Therapy with Kinase Inhibitors Michaela Bairlein Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ. -Prof. Dr. K. Schneitz Prüfer der Dissertation: 1. Univ.-Prof. Dr. A. Gierl 2. Hon.-Prof. Dr. h.c. A. Ullrich (Eberhard-Karls-Universität Tübingen) 3. Univ.-Prof. A. Schnieke, Ph.D. Die Dissertation wurde am 07.01.2010 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 19.04.2010 angenommen. FOR MY PARENTS 1 Contents 2 Summary ................................................................................................................................................................... 5 3 Zusammenfassung .................................................................................................................................................... 6 4 Introduction .............................................................................................................................................................. 8 4.1 Cancer .............................................................................................................................................................. -

Intrinsic Disorder of the BAF Complex: Roles in Chromatin Remodeling and Disease Development
International Journal of Molecular Sciences Article Intrinsic Disorder of the BAF Complex: Roles in Chromatin Remodeling and Disease Development Nashwa El Hadidy 1 and Vladimir N. Uversky 1,2,* 1 Department of Molecular Medicine, Morsani College of Medicine, University of South Florida, 12901 Bruce B. Downs Blvd. MDC07, Tampa, FL 33612, USA; [email protected] 2 Laboratory of New Methods in Biology, Institute for Biological Instrumentation of the Russian Academy of Sciences, Federal Research Center “Pushchino Scientific Center for Biological Research of the Russian Academy of Sciences”, Pushchino, 142290 Moscow Region, Russia * Correspondence: [email protected]; Tel.: +1-813-974-5816; Fax: +1-813-974-7357 Received: 20 September 2019; Accepted: 21 October 2019; Published: 23 October 2019 Abstract: The two-meter-long DNA is compressed into chromatin in the nucleus of every cell, which serves as a significant barrier to transcription. Therefore, for processes such as replication and transcription to occur, the highly compacted chromatin must be relaxed, and the processes required for chromatin reorganization for the aim of replication or transcription are controlled by ATP-dependent nucleosome remodelers. One of the most highly studied remodelers of this kind is the BRG1- or BRM-associated factor complex (BAF complex, also known as SWItch/sucrose non-fermentable (SWI/SNF) complex), which is crucial for the regulation of gene expression and differentiation in eukaryotes. Chromatin remodeling complex BAF is characterized by a highly polymorphic structure, containing from four to 17 subunits encoded by 29 genes. The aim of this paper is to provide an overview of the role of BAF complex in chromatin remodeling and also to use literature mining and a set of computational and bioinformatics tools to analyze structural properties, intrinsic disorder predisposition, and functionalities of its subunits, along with the description of the relations of different BAF complex subunits to the pathogenesis of various human diseases. -

Differential and Overlapping Immune Programs Regulated by IRF3 and IRF5 in Plasmacytoid Dendritic Cells
Differential and Overlapping Immune Programs Regulated by IRF3 and IRF5 in Plasmacytoid Dendritic Cells This information is current as Kwan T. Chow, Courtney Wilkins, Miwako Narita, Richard of September 28, 2021. Green, Megan Knoll, Yueh-Ming Loo and Michael Gale, Jr. J Immunol published online 8 October 2018 http://www.jimmunol.org/content/early/2018/10/05/jimmun ol.1800221 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2018/10/05/jimmunol.180022 Material 1.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 28, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2018 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published October 8, 2018, doi:10.4049/jimmunol.1800221 The Journal of Immunology Differential and Overlapping Immune Programs Regulated by IRF3 and IRF5 in Plasmacytoid Dendritic Cells Kwan T. Chow,*,† Courtney Wilkins,* Miwako Narita,‡ Richard Green,* Megan Knoll,* Yueh-Ming Loo,* and Michael Gale, Jr.* We examined the signaling pathways and cell type–specific responses of IFN regulatory factor (IRF) 5, an immune-regulatory transcription factor. -
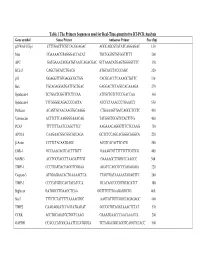
Table 1 the Primers Sequences Used for Real-Time Quantitative RT-PCR
Table 1 The Primers Sequences used for Real-Time quantitative RT-PCR Analysis Gene symbol Sense Primer Antisense Primer Size (bp) p21WAF1/Cip1 CTTTGACTTCGTCACGGAGAC AGGCAGCGTATATCAGGAGAC 150 Max CGAAAACGTAGGGACCACAT TGCTGGTGTGTGGTTTTT 200 APC GATGAAATAGGATGTAATCAGACGAC GCTAAACATGAGTGGGGTCTC 350 BCL-2 CAGCTGCACCTGACG ATGCACCTACCCAGC 320 p53 GGAGGTTGTGAGGCGCTGG CACGCACCTCAAAGCTGTTC 330 Bax TGCAGAGGATGATTGCTGAC GAGGACTCCAGCCACAAAGA 270 Syndecan-1 TCTGACTCGGTTTCTCCAA ATTGCTGTCTCCCGACCAA 360 Syndecan-3 TTTGGGGCAGACCCCACTA ATCCCTAAACCCTGAACCT 550 Perlecan ACAGTGCAACAAGTGCAAGG CTGAAAGTGACCAGGCTCCTC 450 Vitronectin ACTTCTTCAAGGGGAAACAG TATGGCTGCGTCCACTTTG 400 PCAF TTTTTTTAATCCAGCTTCC AAGAAACAGGGTTTCTCCAAG 750 APO1A CAAGAACGGCGGCGCCAGA GCTCTCCAGCACGGGCAGGCA 220 β-Actin CCTTCTACAATGAGC ACGTCACACTTCATG 260 EGR-1 GCCAAACAGTCACTTTGTT GAAAGTGTTTTTTCTTCGTCG 400 MAPK3 ACCTGCGACCTTAAGATTTGT GAAAAGCTTGGCCCAAGCC 300 TIMP-1 CCCTGATGACGAGGTCGGAA AGATCCAGCGCCCAGAGAGA 220 Caspase-3 ATGGAGAACACTGAAAACTCA TTAGTGATAAAAATAGAGTTC 200 TIMP-3 CCCCATGTGCAGTACATCCA GCACAGCCCCGTGTACATCT 180 Biglycan GATGGCCTGAAGCTCAA GGTTTGTTGAAGAGGCTG 460 Stat3 TTTCTCTATTTTTAAAAGTGC AAGTATTGTCGGCCAGAGAGC 460 TIMP2 CAAGAGGATCCAGTATGAGAT GCCCGTGTAGATAAACTCTAT 570 CCRK ACCTGCAGATGCTGCTCAAG GAAATGAACCCAACAAACCA 200 GAPDH CCACCCATGGCAAATTCCATGGGA TCTAGACGGCAGGTCAGGTCCACC 500 Table 2. Genes with Altered Expression in Cleaved vs. Native Antithrombin-Treated HUVECs Gene Fold Changes Gene Symbol Gene Description Cluster ID (ATC/ATN) Adhesion/ECM ARGN Agrin Hs.273330 2.18 CDH24 Caderhrin-like