On the Role of Chromosomal Rearrangements in Evolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Epidemiology
Received: 10 May 2019 | Revised: 31 July 2019 | Accepted: 28 August 2019 DOI: 10.1002/gepi.22260 RESEARCH ARTICLE Population‐wide copy number variation calling using variant call format files from 6,898 individuals Grace Png1,2,3 | Daniel Suveges1,4 | Young‐Chan Park1,2 | Klaudia Walter1 | Kousik Kundu1 | Ioanna Ntalla5 | Emmanouil Tsafantakis6 | Maria Karaleftheri7 | George Dedoussis8 | Eleftheria Zeggini1,3* | Arthur Gilly1,3,9* 1Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton, United Abstract Kingdom Copy number variants (CNVs) play an important role in a number of human 2Department of Medical Genetics, diseases, but the accurate calling of CNVs remains challenging. Most current University of Cambridge, Cambridge, approaches to CNV detection use raw read alignments, which are computationally United Kingdom intensive to process. We use a regression tree‐based approach to call germline CNVs 3Institute of Translational Genomics, Helmholtz Zentrum München—German from whole‐genome sequencing (WGS, >18x) variant call sets in 6,898 samples Research Center for Environmental across four European cohorts, and describe a rich large variation landscape Health, Neuherberg, Germany comprising 1,320 CNVs. Eighty‐one percent of detected events have been previously 4European Bioinformatics Institute, ‐ ‐ Wellcome Genome Campus, Hinxton, reported in the Database of Genomic Variants. Twenty three percent of high quality United Kingdom deletions affect entire genes, and we recapitulate known events such as the GSTM1 5William Harvey Research Institute, Barts and RHD gene deletions. We test for association between the detected deletions and and The London School of Medicine and 275 protein levels in 1,457 individuals to assess the potential clinical impact of the Dentistry, Queen Mary University of London, London, United Kingdom detected CNVs. -

RESOLUCIÓN GRI/3142/2015, De 29 De Diciembre, Por La Que Se Da Publicidad Al Deslinde Entre Los Términos Municipales De Aguilar De Segarra Y De Castellfollit Del Boix
RESOLUCIÓN GRI/3142/2015, de 29 de diciembre, por la que se da publicidad al deslinde entre los términos municipales de Aguilar de Segarra y de Castellfollit del Boix. Organo Emisor: Tipo de Norma: Resolución Fecha: 2015-12-29 12:00:00 Fecha de Publicacion en el BOE: 2016-02-10 12:00:00 Marginal: 69477125 TEXTO COMPLETO : Los días 30 de setiembre de 2013 y 21 de septiembre de 2015, y 23 de octubre de 2013 y 24 de septiembre de 2015, los plenos de los ayuntamientos de Aguilar de Segarra y de Castellfollit del Boix aprobaron, respectivamente, con el voto favorable de la mayoría absoluta del número legal de miembros de la corporación, el deslinde efectuado los días 4 de setiembre de 2013 y 21 de setiembre de 2015 por las respectivas comisiones municipales. Este deslinde forma parte del proceso de elaboración del Mapa municipal de Cataluña, previsto en el artículo 28.1 del Texto refundido de la Ley municipal y de régimen local de Cataluña, aprobado por el Decreto legislativo 2/2003, de 28 de abril. De acuerdo con el artículo 32.1 del Decreto 244/2007, de 6 de noviembre, por el que se regula la constitución y la demarcación territorial de los municipios, de las entidades municipales descentralizadas y de las mancomunidades de Cataluña, modificado por el Decreto 209/2015, de 22 de septiembre, Resuelvo: Dar publicidad al deslinde aprobado por los ayuntamientos de Aguilar de Segarra y de Castellfollit del Boix que se describe a continuación e incorporarlo a los mapas municipales correspondientes. Mojón 1: se sitúa en el lugar conocido como collado de Bolederes, en la divisoria de aguas de la sierra de El Soler. -

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -

Coupling of Spliceosome Complexity to Intron Diversity
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Coupling of spliceosome complexity to intron diversity Jade Sales-Lee1, Daniela S. Perry1, Bradley A. Bowser2, Jolene K. Diedrich3, Beiduo Rao1, Irene Beusch1, John R. Yates III3, Scott W. Roy4,6, and Hiten D. Madhani1,6,7 1Dept. of Biochemistry and Biophysics University of California – San Francisco San Francisco, CA 94158 2Dept. of Molecular and Cellular Biology University of California - Merced Merced, CA 95343 3Department of Molecular Medicine The Scripps Research Institute, La Jolla, CA 92037 4Dept. of Biology San Francisco State University San Francisco, CA 94132 5Chan-Zuckerberg Biohub San Francisco, CA 94158 6Corresponding authors: [email protected], [email protected] 7Lead Contact 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. SUMMARY We determined that over 40 spliceosomal proteins are conserved between many fungal species and humans but were lost during the evolution of S. cerevisiae, an intron-poor yeast with unusually rigid splicing signals. We analyzed null mutations in a subset of these factors, most of which had not been investigated previously, in the intron-rich yeast Cryptococcus neoformans. -
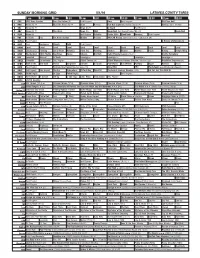
Sunday Morning Grid 5/1/16 Latimes.Com/Tv Times
SUNDAY MORNING GRID 5/1/16 LATIMES.COM/TV TIMES 7 am 7:30 8 am 8:30 9 am 9:30 10 am 10:30 11 am 11:30 12 pm 12:30 2 CBS CBS News Sunday Face the Nation (N) Paid Program Boss Paid Program PGA Tour Golf 4 NBC News (N) Å Meet the Press (N) Å News Rescue Red Bull Signature Series (Taped) Å Hockey: Blues at Stars 5 CW News (N) Å News (N) Å In Touch Paid Program 7 ABC News (N) Å This Week News (N) NBA Basketball First Round: Teams TBA. (N) Basketball 9 KCAL News (N) Joel Osteen Schuller Pastor Mike Woodlands Amazing Paid Program 11 FOX In Touch Paid Fox News Sunday Midday Prerace NASCAR Racing Sprint Cup Series: GEICO 500. (N) 13 MyNet Paid Program A History of Violence (R) 18 KSCI Paid Hormones Church Faith Paid Program 22 KWHY Local Local Local Local Local Local Local Local Local Local Local Local 24 KVCR Landscapes Painting Joy of Paint Wyland’s Paint This Painting Kitchen Mexico Martha Pépin Baking Simply Ming 28 KCET Wunderkind 1001 Nights Bug Bites Space Edisons Biz Kid$ Celtic Thunder Legacy (TVG) Å Soulful Symphony 30 ION Jeremiah Youssef In Touch Leverage Å Leverage Å Leverage Å Leverage Å 34 KMEX Conexión En contacto Paid Program Fútbol Central (N) Fútbol Mexicano Primera División: Toluca vs Azul República Deportiva (N) 40 KTBN Walk in the Win Walk Prince Carpenter Schuller In Touch PowerPoint It Is Written Pathway Super Kelinda Jesse 46 KFTR Paid Program Formula One Racing Russian Grand Prix. -

Gene Expression Imputation Across Multiple Brain Regions Provides Insights Into Schizophrenia Risk
VU Research Portal Gene expression imputation across multiple brain regions provides insights into schizophrenia risk iPSYCH-GEMS Schizophrenia Working Group; CommonMind Consortium; The Schizophrenia Working Group of the PsyUniversity of Copenhagenchiatric Genomics Consortium published in Nature Genetics 2019 DOI (link to publisher) 10.1038/s41588-019-0364-4 document version Publisher's PDF, also known as Version of record document license Article 25fa Dutch Copyright Act Link to publication in VU Research Portal citation for published version (APA) iPSYCH-GEMS Schizophrenia Working Group, CommonMind Consortium, & The Schizophrenia Working Group of the PsyUniversity of Copenhagenchiatric Genomics Consortium (2019). Gene expression imputation across multiple brain regions provides insights into schizophrenia risk. Nature Genetics, 51(4), 659–674. https://doi.org/10.1038/s41588-019-0364-4 General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. E-mail address: [email protected] Download date: 28. -

Objetivo Tren 2024
@transportpublic PromocioTransportPublic @transportpublic www.transportpublic.org [email protected] 93 244 49 70 OBJETIVO TREN 2024 Propuesta ferroviaria para reducir a casi cero las emisiones de CO2 del transporte público en Catalunya / 1a edición: Junio 2020 Autores: Pau Noy Serrano, ingeniero industrial. Ricard Riol Jurado, ingeniero técnico de obras públicas. Colaboradores: Albert Parés i Soldevila, Daniel Pi Noya, Georgina Montesinos Zaragoza, José Luis Rodrigo Jiménez, Josep Maria Olivé i Garcia, Marc Iglesias Pérez, Xavier Lujan Calvo, Javier Paricio Sosa. 2 / 114 ÍNDICE 1. Prólogo ................................................................................................................................ 4 2. ¿Por qué el nuevo Objetivo Tren 2024? ......................................................................... 6 3. ¿Por qué potenciar el ferrocarril? ................................................................................. 14 4. Principios del esquema básico de servicio deseado ................................................... 17 5. Actuaciones prioritarias de infraestructura ................................................................. 21 6. Actuaciones prioritarias no infraestructurales ............................................................ 42 7. Proyectos ferroviarios a estudiar a medio y largo ..................................................... 48 8. Nivel de servicio propuesto para cercanías ................................................................. 53 Conexión ferroviaria del -

L'hospital De Sant Joan Despí Moisès Broggi Inicia Una Campanya De
Col·laboració L’Hospital de Sant Joan Despí Moisès Broggi inicia una campanya de donació de sang setmanal amb el Banc de Sang i Teixits • Es podrà donar sang al centre a partir del 9 de novembre, tots els dimecres laborables de 10 a 13.30 hores L’Hospital de Sant Joan Despí Moisès Broggi i el Banc de Sang i Teixits (BST) han arribat a un acord gràcies al qual, tots els dimecres laborables, a partir del 9 de novembre, tothom que vulgui donar sang a les instal·lacions de l’hospital, ho podrà fer els dimecres de 10 a 13.30 hores, a un espai habilitat a la planta 0, davant de la cafeteria de personal. Fins ara, l’Hospital col·laborava puntualment amb el Banc de Sang i Teixits, amb l’organització de dies de recollida puntual de sang al centre. L’objectiu d’aquesta acció nova és arribar als usuaris que des de fa un any i mig tenen un hospital de referència al seu territori del Baix Llobregat. Són prop de 300.000 persones de Cervelló, Corbera de Llobregat, Cornellà de Llobregat, Esplugues de Llobregat, Molins de Rei, la Palma de Cervelló, el Papiol, Sant Feliu de Llobregat, Sant Joan Despí, Sant Just Desvern i Vallirana. S’espera que aquesta iniciativa, que portarà a la col·laboració estreta entre l’Hospital de Sant Joan Despí Moisès Broggi i el Banc de Sang i Teixitis, sigui ben rebuda i ajudi a obtenir les reserves de sang que necessiten els malalts. El Banc de Sang i Teixits és l'empresa pública del Departament de Salut de la Generalitat de Catalunya que té com a missió garantir l'abastament i el bon ús de sang i teixits a Catalunya, essent el centre de referència en immunologia diagnòstica i de desenvolupament de teràpies avançades. -

A Mutation in the Putative MLH3 Endonuclease Domain Confers a Defect in Both Mismatch Repair and Meiosis in Saccharomyces Cerevisiae
Copyright Ó 2008 by the Genetics Society of America DOI: 10.1534/genetics.108.086645 A Mutation in the Putative MLH3 Endonuclease Domain Confers a Defect in Both Mismatch Repair and Meiosis in Saccharomyces cerevisiae K. T. Nishant, Aaron J. Plys and Eric Alani1 Department of Molecular Biology and Genetics, Cornell University, Ithaca, New York 14853-2703 Manuscript received January 2, 2008 Accepted for publication March 20, 2008 ABSTRACT Interference-dependent crossing over in yeast and mammalian meioses involves the mismatch repair protein homologs MSH4-MSH5 and MLH1-MLH3. The MLH3 protein contains a highly conserved metal- binding motif DQHA(X)2E(X)4E that is found in a subset of MLH proteins predicted to have endonuclease activities (Kadyrov et al. 2006). Mutations within this motif in human PMS2 and Saccharomyces cerevisiae PMS1 disrupted the endonuclease and mismatch repair activities of MLH1-PMS2 and MLH1-PMS1, re- spectively (Kadyrov et al. 2006, 2007; Erdeniz et al. 2007). As a first step in determining whether such an activity is required during meiosis, we made mutations in the MLH3 putative endonuclease domain motif (-D523N, -E529K) and found that single and double mutations conferred mlh3-null-like defects with respect to meiotic spore viability and crossing over. Yeast two-hybrid and chromatography analyses showed that the interaction between MLH1 and mlh3-D523N was maintained, suggesting that the mlh3-D523N mutation did not disrupt the stability of MLH3. The mlh3-D523N mutant also displayed a mutator phenotype in vegetative growth that was similar to mlh3D. Overexpression of this allele conferred a dominant-negative phenotype with respect to mismatch repair. -

Ck1δ Over-Expressing Mice Display ADHD-Like Behaviors, Frontostriatal Neuronal Abnormalities and Altered Expressions of ADHD-Candidate Genes
Molecular Psychiatry (2020) 25:3322–3336 https://doi.org/10.1038/s41380-018-0233-z ARTICLE CK1δ over-expressing mice display ADHD-like behaviors, frontostriatal neuronal abnormalities and altered expressions of ADHD-candidate genes 1 1 1 2 1 1 1 Mingming Zhou ● Jodi Gresack ● Jia Cheng ● Kunihiro Uryu ● Lars Brichta ● Paul Greengard ● Marc Flajolet Received: 8 November 2017 / Revised: 4 July 2018 / Accepted: 18 July 2018 / Published online: 19 October 2018 © Springer Nature Limited 2018 Abstract The cognitive mechanisms underlying attention-deficit hyperactivity disorder (ADHD), a highly heritable disorder with an array of candidate genes and unclear genetic architecture, remain poorly understood. We previously demonstrated that mice overexpressing CK1δ (CK1δ OE) in the forebrain show hyperactivity and ADHD-like pharmacological responses to D- amphetamine. Here, we demonstrate that CK1δ OE mice exhibit impaired visual attention and a lack of D-amphetamine- induced place preference, indicating a disruption of the dopamine-dependent reward pathway. We also demonstrate the presence of abnormalities in the frontostriatal circuitry, differences in synaptic ultra-structures by electron microscopy, as 1234567890();,: 1234567890();,: well as electrophysiological perturbations of both glutamatergic and GABAergic transmission, as observed by altered frequency and amplitude of mEPSCs and mIPSCs. Furthermore, gene expression profiling by next-generation sequencing alone, or in combination with bacTRAP technology to study specifically Drd1a versus Drd2 medium spiny neurons, revealed that developmental CK1δ OE alters transcriptional homeostasis in the striatum, including specific alterations in Drd1a versus Drd2 neurons. These results led us to perform a fine molecular characterization of targeted gene networks and pathway analysis. Importantly, a large fraction of 92 genes identified by GWAS studies as associated with ADHD in humans are significantly altered in our mouse model. -

7.6.5 Àmbit Del Pla Del Bages
7.6.5 Àmbit del Pla del Bages Descripció de l’àmbit L’àmbit té una extensió de 985 km2 i compren la major part de la comarca ubicada al bell mig de la Depressió Central Catalana. Està constituït per 27 municipis: Sant Mateu de Bages, Aguilar de Segarra i Fonollosa, al NW del sistema, participen de l’altiplà segarrenc; Santa Maria d’Oló és localitza limitant amb Osona entre els altiplans del Lluçanès i del Moianès; El Pont de Vilomara i Rocafort, Sant Vicenç de Castellet, Castellbell i el Vilar, Monistrol de Montserrat i Marganell localitzats a la part sud-oriental de l’àmbit es situen sobre la Serralada Prelitoral; la resta de municipis - Artés, Avinyó, Balsareny, Callús, Castellfollit del Boix, Castellgalí, Castellnou de Bages, Gaià, Manresa, Navarcles, Navàs, Rajadell, Sallent, Sant Fruitós de Bages, Sant Joan de Vilatorrada, Sant Salvador de Guardiola, Santpedor i Súria -, constitueixen pròpiament el pla del Bages que està format per una gran conca d’erosió. El sistema d’ assentaments 7-50 El riu Llobregat travessa l’àmbit en direcció N-S i l’abandona pel congost de Monistrol. Al Sud de Manresa hi conflueix el Cardener, un dels seus afluents més importants que creua el sector en direcció NW. En conjunt, el 22% de la superfície de l’àmbit és planer, el 28% té un pendent comprès entre el 10 i el 20%, i el 50% té un pendent superior al 20%. El sistema urbà que es desenvolupa a l’àmbit del Pla de Bages, més extens que el definit en termes geogràfics, es caracteritza per les evidents continuïtats dels assentaments i implantacions industrials. -

Reference Gene Validation Via RT–Qpcr for Human Ipsc-Derived Neural Stem Cells and Neural Progenitors
Molecular Neurobiology https://doi.org/10.1007/s12035-019-1538-x Reference Gene Validation via RT–qPCR for Human iPSC-Derived Neural Stem Cells and Neural Progenitors Justyna Augustyniak1 & Jacek Lenart2 & Gabriela Lipka1 & Piotr P. Stepien3 & Leonora Buzanska1 Received: 26 November 2018 /Accepted: 22 February 2019 # The Author(s) 2019 Abstract Correct selection of the reference gene(s) is the most important step in gene expression analysis. The aims of this study were to identify and evaluate the panel of possible reference genes in neural stem cells (NSC), early neural progenitors (eNP) and neural progenitors (NP) obtained from human-induced pluripotent stem cells (hiPSC). The stability of expression of genes commonly used as the reference in cells during neural differentiation is variable and does not meet the criteria for reference genes. In the present work, we evaluated the stability of expression of 16 candidate reference genes using the four most popular algorithms: the ΔCt method, BestKeeper, geNorm and NormFinder. All data were analysed using the online tool RefFinder to obtain a comprehensive ranking. Our results indicate that NormFinder is the best tool for reference gene selection in early stages of hiPSC neural differentiation. None of the 16 tested genes is suitable as reference gene for all three stages of development. We recommend using different genes (panel of genes) to normalise RT–qPCR data for each of the neural differentiation stages. Keywords human induced Pluripotent Stem Cell (hiPSC) . Neural Stem Cells . Neural