Introduction to Microbiology Biosci 221 Laboratory Manual
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rapid Identification of Clinical Isolates of Klebsiella Pneumoniae Using MALDI-TOF MS from North India
Bulletin of Pure and Applied Sciences Print version ISSN 0970 0765 Vol.39A (Zoology), No.1, Online version ISSN 2320 3188 January-June 2020: P.194-199 DOI 10.5958/2320-3188.2020.00022.4 Original Research Article Available online at www.bpasjournals.com Rapid Identification of Clinical Isolates of Klebsiella pneumoniae using MALDI-TOF MS from North India 1Sunil Kumar Abstract: 2Zainab Saifi Infections caused by multi-drug resistant (MDR) 3Anil Kumar Sharma Klebsiella pneumoniae are increasing day by day. K. pneumoniae poses a severe public health concern 4 * Sushil Kumar Upadhyay and causes wide array of healthcare associated infections with limited treatment options. The Author’s Affiliation: quick detection of these isolates is of prime 1,2,3,4 Department of Biotechnology, Maharishi importance for the adoption of proper antibiotic Markandeshwar (Deemed to be University), treatment and control measures. Also the mis- Mullana-Ambala, Haryana 133207, India identification using standard lab-based methods is quite common and hence, the clinical *Corresponding author: significance of K. pneumoniae complex members is Dr. Sushil Kumar Upadhyay, inaccurately defined. Here, authors evaluated the Assistant Professor, Department of potential of MALDI-TOF (Matrix-Assisted Laser Biotechnology, Maharishi Markandeshwar Desorption Ionization-Time of Flight) MS (Mass (Deemed to be University), Mullana-Ambala, Spectrometry) to discriminate and quick Haryana 133207, India identification K. pneumoniae complex members. Thus, this study aimed at full MALDI-based E-mail: approach to rapidly detect the K. [email protected] pneumoniae isolates up to species level. We also ORCID: https://orcid.org/0000-0002-1229-4275 explored the antibiotic resistance profile of K. -

Characterization and Antibiotic Sensitivity Profile of Bacteria Isolated from Patients with Respiratory Tract Infections in Bangladesh
Characterization and Antibiotic Sensitivity Profile of Bacteria Isolated from Patients with Respiratory Tract Infections in Bangladesh Shukla Promite1, Sajal K. Saha2, Sunjukta Ahsan1 and Marufa Zerin Akhter1 1Department of Microbiology, University of Dhaka, Dhaka, Bangladesh 2Department of General Practice, Monash University, Building 1, 270 Ferntree Gully Road, Notting Hill VIC 3168, Australia (Received: October 08, 2017; Accepted: December 15, 2017; Published (web): December 23, 2017) ABSTRACT: The study was aimed to characterize bacterial isolates from respiratory tract infections (RTI) and investigate their antibiotic sensitivity profile. Selective media and biochemical tests were used to characterize 40 bacterial isolates. Antibiotic sensitivity testing was conducted using Kirby-Bauer disc diffusion method. About 42.5% (17) RTI patients were infected by Klebsiella pneumoniae, 30% (12) by Escherichia coli and 27.5% (11) by Pseudomonas aeruginosa with no significant gender variation (p-value <0.578). Overall, 47% (out of 20) antibiotics were sensitive, whereas 48% were resistant. Surprisingly, 18% P. aeruginosa and 20% K. pneumoniae were carbapenem-resistant and 4 out of 7 cephalosporin antibiotics were highly resistant irrespective of pathogens. E. coli showed better sensitivity to nitrofurantoin (78%) and levofloxacin (89%), while K. pneumoniae was insensitive to cotrimoxazole (88%), gentamycin (77%) and piperacillin/tazobactam (66%). On the other hand, P. aeruginosa did not respond to P. aeruginosa to nalidixic acid (60%) and ciprofloxacin (60%). This study concludes that nitrofurantoin, levofloxacin, cotrimoxazole, gentamycin and piperacillin/tazobactam antibiotics could be better alternative in treating bacterial RTIs. Key words: Antibiotic sensitivity, bacterial pathogens, RTIs, Bangladesh. INTRODUCTION Antibiotic resistance (AR) is a global public The rise of AR in Bangladesh is probably due to 1 health concern. -

Npgrj Nprot 406 2517..2526
PROTOCOL Identification and analysis of essential Aspergillus nidulans genes using the heterokaryon rescue technique Aysha H Osmani, Berl R Oakley & Stephen A Osmani Department of Molecular Genetics, The Ohio State University, Columbus, Ohio 43210, USA. Correspondence should be addressed to S.A.O. ([email protected]) Published online 29 December 2006; corrected online 25 January 2007 (details online); doi:10.1038/nprot.2006.406 s In the heterokaryon rescue technique, gene deletions are carried out using the pyrG nutritional marker to replace the coding region of target genes via homologous recombination in Aspergillus nidulans. If an essential gene is deleted, the null allele is maintained in spontaneously generated heterokaryons that consist of two genetically distinct types of nuclei. One nuclear type has the essential gene deleted but has a functional pyrG allele (pyrG+). The other has the wild-type allele of the essential gene but lacks a functional pyrG allele (pyrG–). Thus, a simple growth test applied to the uninucleate asexual spores formed from primary transformants can natureprotocol / m identify deletions of genes that are non-essential from those that are essential and can only be propagated by heterokaryon rescue. o c . The growth tests also enable the phenotype of the null allele to be defined. Diagnostic PCR can be used to confirm deletions at the e r molecular level. This technique is suitable for large-scale gene-deletion programs and can be completed within 3 weeks. u t a n . w w INTRODUCTION w / / : One of the most fundamental pieces of information regarding the non-essential gene is deleted, the resulting strains are able to grow p t t function of any gene is whether the gene is essential or not. -

Microbiology Laboratory Exercises Third Edition 2020
MICROBIOLOGY Laboratory Exercises Third Edition Keddis & Rauschenbach 2020 Photo Credits (in order of contribution): Diane Davis, Ines Rauschenbach & Ramaydalis Keddis Acknowledgements: Many thanks to those in the Department of Biochemistry and Microbiology, Rutgers University, who have through the years inspired our enthusiasm for the science and teaching of microbiology, with special thanks to Diane Davis, Douglas Eveleigh and Max Häggblom. Safety: The experiments included in this manual have been deemed safe by the authors when all necessary safety precautions are met. The authors recommend maintaining biosafety level 2 in the laboratory setting and using risk level 1 organisms for all exercises. License: This work is licensed under a Creative Commons Attribution- NonCommercial-NoDerivatives 4.0 International License Microbiology Laboratory Exercises Third Edition 2020 Ramaydalis Keddis, Ph.D. Ines Rauschenbach, Ph.D. Department of Biochemistry and Microbiology Rutgers, The State University of New Jersey CONTENTS PAGE Introduction Schedule ii Best Laboratory Practices Iii Working in a Microbiology Laboratory iv Exercises Preparation of a Culture Medium 1 Culturing and Handling Microorganisms 3 Isolation of a Pure Culture 5 Counting Bacterial Populations 8 Controlling Microorganisms 10 Disinfectants 10 Antimicrobial Agents: Susceptibility Testing 12 Hand Washing 14 The Lethal Effects of Ultraviolet Light 15 Selection of Fungi from Air 17 Microscopy 21 Morphology and Staining of Bacteria 26 Microbial Metabolism 30 Enzyme Assay 32 Metabolic -
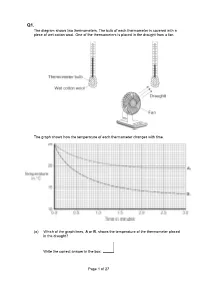
Page 1 of 27 the Diagram Shows Two Thermometers. the Bulb of Each
Q1. The diagram shows two thermometers. The bulb of each thermometer is covered with a piece of wet cotton wool. One of the thermometers is placed in the draught from a fan. The graph shows how the temperature of each thermometer changes with time. (a) Which of the graph lines, A or B, shows the temperature of the thermometer placed in the draught? Write the correct answer in the box. Page 1 of 27 Explain, in terms of evaporation, the reason for your answer. ___________________________________________________________________ ___________________________________________________________________ ___________________________________________________________________ ___________________________________________________________________ ___________________________________________________________________ ___________________________________________________________________ (3) (b) A wet towel spread out and hung outside on a day without wind dries faster than an identical wet towel left rolled up in a plastic bag. Explain why. ___________________________________________________________________ ___________________________________________________________________ ___________________________________________________________________ ___________________________________________________________________ (2) (Total 5 marks) Q2. The picture shows a person taking a hot shower. (a) When a person uses the shower the mirror gets misty. Why? ___________________________________________________________________ ___________________________________________________________________ -

Pasteurella Multocida Isolated from Cattle
Journal of Applied Pharmaceutical Science Vol. 3 (04), pp. 106-110, April, 2013 Available online at http://www.japsonline.com DOI: 10.7324/JAPS.2013.3419 ISSN 2231-3354 Antibiotic Susceptibility and Molecular Analysis of Bacterial Pathogen Pasteurella Multocida Isolated from Cattle Azmat Jabeen, Mahrukh Khattak, Shahzad Munir*, Qaiser Jamal, Mubashir Hussain Department of Microbiology, Kohat University of Science and Technology, Kohat, Khyber Pakhtunkhwa, Pakistan. ARTICLE INFO ABSTRACT Article history: Pasteurella multocida is a Gram negative, non motile and coccobacillus bacterium. It has 5 strains i.e. A, B, D, Received on: 01/02/2013 E and F and 16 serotypes (1-16). In present study, we analyzed Pasteurella multocida B: 2 strains, responsible Revised on: 19/02/2013 for Hemorrhagic Septicemia (HS) in cattle, on morphological/microbial, biochemical, molecular level and to Accepted on: 15/03/2013 check the antibiotic sensitivity of the Pasteurella multocida. Microbial analysis showed that while grown on Available online: 27/04/2013 Brain Heart Infusion agar plates and Blood Agar Base Medium, grayish lustrous colonies of Pasteurella multocida were observed. Gram staining showed that Pasteurella multocida are gram negative. Microscopic Key words: observations revealed it to be coccobacillus and it was non- motile. Identification was conducted by Pasteurella multocida, conventional biochemical tests and percentage identification of Analytical Profile Index was 96 %. Antibiotic Hemorrhagic Septicemia, sensitivity with different antibiotics was checked by disk diffusion method and was found resistant to Analytical Profile Index, Augmentin, Amoxicillin and Aztreonam and was more susceptible to Ceftiofur. On molecular level its DNA Antibiotic sensitivity. was extracted and was run with marker having range from 0.5 – 10 kb. -

Harvard Biolabs Stockroom
THE HARVARD BIOLABS STOCKROOM Biolabs Basement—B060 Biolabs Bldg ‐ 16 Divinity Ave Phone: 617.495.2385 Monday ‐ Friday: 8:00 am ‐ 4:50 pm* *Closed daily 12:00 ‐ 1:00 pm The Harvard Biolabs Stockroom The Harvard BioLabs Stockroom Biolabs Basement—B060 The current Harvard Biolabs Stockroom was created in collaboraon with Operaons and Facilies at FAS and VWR Internaonal to provide researchers with quick, convenient access to the most frequently ordered laboratory supplies, enzymes and biologicals. Products are sourced from VWR core suppliers, as well as other popular manufacturers such as Qiagen, NEB, Falcon and Corning. Researchers using Harvard funds for payment are eligible to purchase from the Stockroom. To ensure accurate billing, shoppers should be prepared to provide idenficaon and up‐to‐date grant informaon at the request of Stockroom personnel. All non‐stockroom orders should be placed on‐line via HCOM or by calling VWR at 866‐229‐9967 “Call Ahead” ‐The Stockroom offers walk‐ in service. However, you may want to “call ahead” to 617‐495‐2385 so the VWR associate can ensure availability of the products requested. VWR will provide a receipt detailing all items on the order. Only products in stock will be included. Back orders will not be accepted. Backordered products should be ordered as a new transacon when stock arrives. Check with VWR at 866‐229‐9967 or HCOM for availability. VWR manages the 190 and 200 proof tax free ethanol program for Harvard University. Ethanol can be purchased by the gallon(s) or pint in the Stockroom. The on‐campus stockroom is intended to meet immediate needs for less‐ than‐case quanty products; for larger orders it is recommended purchases be made on line via HCOM, by calling 866‐229‐9967 or by e‐mailing [email protected]. -

Pouring Plates from Prepared Bottled Media
Pouring Plates from Prepared Bottled Media Primary Hazard Warning Never purchase living specimens without having a disposition strategy in place. When pouring bottles, agar is HOT! Burning can occur. Always handle hot agar bottles with heat-protective gloves. For added protection wear latex or nitrile gloves when working with bacteria, and always wash hands before and after with hot water and soap. Availability Agar is available for purchase year round. Information • Storage: Bottled agar can be stored at room temperature for about six months unless otherwise specified. Never put agar in the freezer. It will cause the agar to breakdown and become unusable. To prevent contamination keep all bottles and Petri dishes sealed until ready to use. • Pouring Plates • Materials Needed: • Draft-free enclosure or Laminar flow hood • 70% isopropyl alcohol • Petri dishes • Microwave or hot water bath or autoclave 1. Melt the agar using one of the following methods: a) Autoclave: Loosen the cap on the agar bottle and autoclave the bottle at 15 psi for five minutes. While wearing heat-protective gloves, carefully remove the hot bottle and let it cool to between 75–55°C before pouring. This takes approximately 15 minutes. b)Water Bath: Loosen the cap on the agar bottle and place it into a water bath. Water temperature should remain at around 100°C. Leave it in the water bath until the agar is completely melted. While wearing heat- protective gloves, carefully remove the hot bottle and let it cool to between 75–55°C before pouring. c) Microwave: Loosen the cap on the agar bottle before microwaving. -

Identification of Pasteurella Species and Morphologically Similar Organisms
UK Standards for Microbiology Investigations Identification of Pasteurella species and Morphologically Similar Organisms Issued by the Standards Unit, Microbiology Services, PHE Bacteriology – Identification | ID 13 | Issue no: 3 | Issue date: 04.02.15 | Page: 1 of 28 © Crown copyright 2015 Identification of Pasteurella species and Morphologically Similar Organisms Acknowledgments UK Standards for Microbiology Investigations (SMIs) are developed under the auspices of Public Health England (PHE) working in partnership with the National Health Service (NHS), Public Health Wales and with the professional organisations whose logos are displayed below and listed on the website https://www.gov.uk/uk- standards-for-microbiology-investigations-smi-quality-and-consistency-in-clinical- laboratories. SMIs are developed, reviewed and revised by various working groups which are overseen by a steering committee (see https://www.gov.uk/government/groups/standards-for-microbiology-investigations- steering-committee). The contributions of many individuals in clinical, specialist and reference laboratories who have provided information and comments during the development of this document are acknowledged. We are grateful to the Medical Editors for editing the medical content. For further information please contact us at: Standards Unit Microbiology Services Public Health England 61 Colindale Avenue London NW9 5EQ E-mail: [email protected] Website: https://www.gov.uk/uk-standards-for-microbiology-investigations-smi-quality- and-consistency-in-clinical-laboratories UK Standards for Microbiology Investigations are produced in association with: Logos correct at time of publishing. Bacteriology – Identification | ID 13 | Issue no: 3 | Issue date: 04.02.15 | Page: 2 of 28 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Identification of Pasteurella species and Morphologically Similar Organisms Contents ACKNOWLEDGMENTS ......................................................................................................... -

Microlab® STAR™
Microlab ® STAR™ Microlab ® STAR ™ AUTOMATED WORKFLOW SOLUTIONS CENTERED AROUND YOUR ASSAY The STAR combines Hamilton's patented pipetting technology including precise lock-and-key tip attachment, unrivaled liquid level detection, and comprehensive volume ranges to create flexible liquid handling workstations. Available in three base platform sizes, the STAR portfolio incorporates countless options to automate your workflows. Hamilton Robotics has also partnered with top leaders in the biotechnology industry to provide Standard Solutions based on commonly automated applications. Offering ready-to-start protocols for a variety of applications such as NGS, ELISA, and forensic assays, our Standard Solutions provide a faster way to automate your processes. 2 1 PATENTED TECHNOLOGY The STAR utilizes Hamilton’s proprietary Compressed O-Ring Expansion (CO-RE®) technology. CO-RE minimizes the production of aerosols and allows disposable tips or washable, steel needles to be used on channels in the same run. 2 MULTI-FUNCTIONAL ARM Our technology offers high pipetting accuracy and precision, from sub-microliter to large volumes, using Independent Channels and/or the Multi-Probe Head (MPH). Labware transportation is possible with the iSWAP® or CO-RE Grippers. The STAR can incorporate a camera, tube transportation, and other channel tools on a single arm. Comprehensive pipetting range: ■■■0.5 μL to 1 mL using the 1 mL Independent Channel ■■■50 μL to 5 mL using the 5 mL Independent Channel ■■■1 μL to 1 mL using the CO-RE 96 MPH ■■■0.1 μL to 50 μL using the CO-RE 384 MPH 3 FLEXIBLE SETUP The high-capacity deck is customized specific to your workflow, accommodating a wide range of labware and automated devices that can easily be exchanged to support multiple assays on one platform. -

Bacterial Survival in Microscopic Surface Wetness Maor Grinberg†, Tomer Orevi†, Shifra Steinberg, Nadav Kashtan*
RESEARCH ARTICLE Bacterial survival in microscopic surface wetness Maor Grinberg†, Tomer Orevi†, Shifra Steinberg, Nadav Kashtan* Department of Plant Pathology and Microbiology, Robert H. Smith Faculty of Agriculture, Food, and Environment, Hebrew University, Rehovot, Israel Abstract Plant leaves constitute a huge microbial habitat of global importance. How microorganisms survive the dry daytime on leaves and avoid desiccation is not well understood. There is evidence that microscopic surface wetness in the form of thin films and micrometer-sized droplets, invisible to the naked eye, persists on leaves during daytime due to deliquescence – the absorption of water until dissolution – of hygroscopic aerosols. Here, we study how such microscopic wetness affects cell survival. We show that, on surfaces drying under moderate humidity, stable microdroplets form around bacterial aggregates due to capillary pinning and deliquescence. Notably, droplet-size increases with aggregate-size, and cell survival is higher the larger the droplet. This phenomenon was observed for 13 bacterial species, two of which – Pseudomonas fluorescens and P. putida – were studied in depth. Microdroplet formation around aggregates is likely key to bacterial survival in a variety of unsaturated microbial habitats, including leaf surfaces. DOI: https://doi.org/10.7554/eLife.48508.001 Introduction *For correspondence: The phyllosphere – the aerial parts of plants – is a vast microbial habitat that is home to diverse [email protected] microbial communities (Lindow and Brandl, 2003; Lindow and Leveau, 2002; Vorholt, 2012; Vacher et al., 2016; Leveau, 2015; Bringel and CouA˜ ce, 2015). These communities, dominated by †These authors contributed bacteria, play a major role in the function and health of their host plant, and take part in global bio- equally to this work geochemical cycles. -

BBL Group a Selective Strep Agar with 5% Sheep Blood (Ssa )
BBL™ Group A Selective Strep Agar with 5% Sheep Blood (ssA™) and ! BBL™ Trypticase™ Soy Agar with 5% Sheep Blood (TSA II)–Bi-Plate " L007379 • Rev. 09 • December 2006 QUALITY CONTROL PROCEDURES I INTRODUCTION Group A Selective Strep Agar with 5% Sheep Blood (ssA) is a selective medium for use in the isolation and presumptive identification of group A streptococci from throat cultures and other specimens. Trypticase Soy Agar with 5% Sheep Blood (TSA II) is used for the growth of fastidious organisms and for the visualization of hemolytic reactions. The TSA II medium sector is marked "I" and the ssA medium sector is marked "II" in the bi-plate dish. II PERFORMANCE TEST PROCEDURE A. Group A Selective Strep Agar with 5% Sheep Blood 1. Inoculate representative samples with the cultures diluted to contain 103–104 CFU/0.01 mL. a. To each plate, add 0.01 mL of the dilution and streak for isolation. Make a stab in the primary streak area before streaking the rest of the plate. b. Place a Taxo™ A disc at the intersection of the first and second area of streaking on all plates inoculated with S. pyogenes strains. c. Incubate plates at 35 ± 2°C in an aerobic atmosphere supplemented with carbon dioxide. d. Include Trypticase Soy Agar with 5% Sheep Blood (TSA II) plates as nonselective controls for all organisms. 2. Examine plates after 18–24 h for beta hemolysis in the stabbed area and for amount of growth, inhibition, colony size and hemolytic reactions. Read and record the size of the zone around the Taxo A disc with S.