The Crystal Structure and Mechanism of Orotidine 5-Monophosphate Decarboxylase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metabolite Name Chemical Formula KEGG/HMDB/ Pubchem Identifier Glyoxylate C2H2O3 C00048 Glycolate C2H4O3 C00160 Pyruvate C3H4O3
Metabolite name Chemical formula KEGG/HMDB/ PubChem Identifier glyoxylate C2H2O3 C00048 glycolate C2H4O3 C00160 pyruvate C3H4O3 C00022 lactate C3H6O3 C00186 2-oxobutanoate C4H6O3 C00109 acetoacetate C4H6O3 C00164 glycerate C3H6O4 C00258 uracil C4H4N2O2 C00106 fumarate C4H4O4 C00122 Maleic acid C4H4O4 C01384 2-keto-isovalerate C5H8O3 C00141 Guanidoacetic acid C3H7N3O2 C00581 succinate C4H6O4 C00042 Methylmalonic acid C4H6O4 C02170 3-S-methylthiopropionate C4H8O2S C08276 nicotinate C6H5NO2 C00253 taurine C2H7NO3S C00245 Pyroglutamic acid C5H7NO3 C01879 Citraconic acid C5H6O4 C02226 2-ketohaxanoic acid C6H10O3 HMDB01864 N-Acetyl-L-alanine C5H9NO3 C01073 oxaloacetate C4H4O5 C00036 Hydroxyisocaproic acid C6H12O3 HMDB00746 malate C4H6O5 C00149 hypoxanthine C5H4N4O C00262 anthranilate C7H7NO2 C00108 p-aminobenzoate C7H7NO2 C00568 p-hydroxybenzoate C7H6O3 C00156 acetylphosphate C2H5O5P C00227 Carbamoyl phosphate CH4NO5P C00169 a-ketoglutarate C5H6O5 C00026 Phenylpropiolic acid C9H6O2 HMDB00563 2-oxo-4-methylthiobutanoate C5H8O3S C01180 2-Hydroxy-2-methylbutanedioic acid C5H8O5 C02612 3-methylphenylacetic acid C9H10O2 HMDB02222 xanthine C5H4N4O2 C00385 Hydroxyphenylacetic acid C8H8O3 C05852 2,3-dihydroxybenzoic acid C7H6O4 C00196 orotate C5H4N2O4 C00295 dihydroorotate C5H6N2O4 C00337 allantoin C4H6N4O3 C01551 Aminoadipic acid C6H11NO4 C00956 Indole-3-carboxylic acid C9H7NO2 HMDB03320 phenylpyruvate C9H8O3 C00166 Atrolactic acid C9H10O3 HMDB00475 Phenyllactic acid C9H10O3 C01479 quinolinate C7H5NO4 C03722 phosphoenolpyruvate C3H5O6P C00074 Uric -
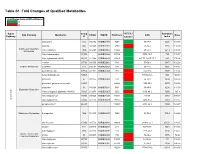
Table S1. Fold Changes of Qualified Metabolites
Table S1. Fold Changes of Qualified Metabolites Fold Change: Cases of WTC-LI/Control <1 >1 Super Comp WTC-LI Retention Sub Pathway Metabolite KEGG HMDB PubChem CAS Mass Pathway ID Control Index asparagine 512 C00152 HMDB00168 6267 0.99 70-47-3 1225 133.06 aspartate 443 C00049 HMDB00191 5960 1.09 56-84-8 1165 134.04 Alanine and Aspartate N-acetylalanine 1585 C02847 HMDB00766 88064 0.99 97-69-8 861.2 130.05 Metabolism N-acetylasparagine 33942 HMDB06028 99715 0.82 4033-40-3 785 175.07 N-acetylaspartate (NAA) 22185 C01042 HMDB00812 65065 0.95 997-55-7;997-55-7 880 176.06 creatine 27718 C00300 HMDB00064 586 0.95 57-00-1 1947 132.08 Creatine Metabolism creatinine 513 C00791 HMDB00562 588 0.99 60-27-5 2055 114.07 guanidinoacetate 43802 C00581 HMDB00128 763 1.05 352-97-6 1937 118.06 beta-citrylglutamate 54923 1.05 73590-26-8 900 322.08 glutamate 57 C00025 HMDB00148 611 1.06 56-86-0 1500 148.06 glutamate, gamma-methyl ester 33487 68662 0.89 1499-55-4 2170 162.08 glutamine 53 C00064 HMDB00641 5961 0.89 56-85-9 1291 147.08 Glutamate Metabolism N-acetyl-aspartyl-glutamate (NAAG) 35665 C12270 HMDB01067 5255 1.04 3106-85-2 1035 305.1 N-acetylglutamate 15720 C00624 HMDB01138 70914 0.84 8/3/17 1050 190.07 Amino Acid Amino N-acetylglutamine 33943 C02716 HMDB06029 182230 0.48 2490-97-3 2140 187.07 pyroglutamine* 46225 134508 0.89 2353-44-8 1900 129.07 Glutathione Metabolism 5-oxoproline 1494 C01879 HMDB00267 7405 0.93 98-79-3 738.5 128.04 allo-threonine 15142 C05519 HMDB04041 99289 0.91 28954-12-3 2511.1 118.05 betaine 3141 C00719 HMDB00043 247 0.92 107-43-7 -

ACS Style Guide
➤ ➤ ➤ ➤ ➤ The ACS Style Guide ➤ ➤ ➤ ➤ ➤ THIRD EDITION The ACS Style Guide Effective Communication of Scientific Information Anne M. Coghill Lorrin R. Garson Editors AMERICAN CHEMICAL SOCIETY Washington, DC OXFORD UNIVERSITY PRESS New York Oxford 2006 Oxford University Press Oxford New York Athens Auckland Bangkok Bogotá Buenos Aires Calcutta Cape Town Chennai Dar es Salaam Delhi Florence Hong Kong Istanbul Karachi Kuala Lumpur Madrid Melbourne Mexico City Mumbai Nairobi Paris São Paulo Singapore Taipei Tokyo Toronto Warsaw and associated companies in Berlin Idaban Copyright © 2006 by the American Chemical Society, Washington, DC Developed and distributed in partnership by the American Chemical Society and Oxford University Press Published by Oxford University Press, Inc. 198 Madison Avenue, New York, NY 10016 Oxford is a registered trademark of Oxford University Press All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, or otherwise, without the prior permission of the American Chemical Society. Library of Congress Cataloging-in-Publication Data The ACS style guide : effective communication of scientific information.—3rd ed. / Anne M. Coghill [and] Lorrin R. Garson, editors. p. cm. Includes bibliographical references and index. ISBN-13: 978-0-8412-3999-9 (cloth : alk. paper) 1. Chemical literature—Authorship—Handbooks, manuals, etc. 2. Scientific literature— Authorship—Handbooks, manuals, etc. 3. English language—Style—Handbooks, manuals, etc. 4. Authorship—Style manuals. I. Coghill, Anne M. II. Garson, Lorrin R. III. American Chemical Society QD8.5.A25 2006 808'.06654—dc22 2006040668 1 3 5 7 9 8 6 4 2 Printed in the United States of America on acid-free paper ➤ ➤ ➤ ➤ ➤ Contents Foreword. -

Biosynthesis of Bromegrass Mosaic Virus Ribonucleic Acid by Dean
Biosynthesis of bromegrass mosaic virus ribonucleic acid by Dean Russell Branson A thesis submitted to the Graduate Faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in BOTANY Montana State University © Copyright by Dean Russell Branson (1967) Abstract: Both de novo synthesis of nucleotides and rearrangement of ribosomal breakdown products appear to be precursor sources for bromegrass mosaic virus (BMV)-RNA. The incorporation of C^14-orotic acid and C^14-uridine, intermediates of the de novo synthesis and breakdown pathways respectively, were compared in BMV-infected barley during a period of virus synthesis. Analyses of the C^14-BMV-RNA specific activities using the dilution factor method showed that the carbon-14 from C^14-uridine was incorporated into BMV-RNA two to three times more efficiently than the carbon-14 from C^14-orotic acid. The essential enzymes for both pathways were demonstrated by using crude enzyme preparations. Those enzymes considered essential were: 1) uridine 5'kinase, 2) orotidine 5’phosphate pyrophosphorylase, and 3) orotidine 5'phosphate decarboxylase. Ribosomal content was lower in BMV-infected tissue than in non-infected tissue suggesting that BMV incorporated the products of host-RNA breakdown, in a manner similar to that reported for tobacco mosaic virus infections. Continuation of ribosomal synthesis in virus infected tissue was demonstrated by the incorporation of C^14-orotic acid into ribosomal RNA. Pyrimidine metabolism, involved in the formation of RNA precursors in BMV-infected and non-infected barley, appeared unchanged except for the additional function of BMV-RNA synthesis. Evidence for this unchanged metabolism was: 1) radioactive ribosomal RNA from C^14-orotic acid-fed barley, infected and non-infected, contained C^14-uridylic acid 3' and C^14-cytidylic acid 3’ when acid hydrolysed, and 2) ribosome breakdown was not a prerequisite for BMV-RNA precursor formation. -
Effects of 6-Azauridine on Nucleotides, Orotic Acid, and Orotidine in L5178Y Mouse Lymphoma Cells in Wfro1
[CANCER RESEARCH 37, 4382-4388, December 1977] Effects of 6-Azauridine on Nucleotides, Orotic Acid, and Orotidine in L5178Y Mouse Lymphoma Cells in Wfro1 Claude M. Janeway and Sungman Cha Division ot Biology and Medicine, Brown University, Providence, Rhode Island 02912 SUMMARY In addition to drastic changes in pyrimidine nucleotides and their precursors, significant effects have been observed Murine lymphoma L5178Y cells in culture were exposed in the purine nucleotide concentrations. A working hypoth to 5 fj.M 6-azauridine (6-aza-UR) for up to 8 hr. The nucleo- esis is offered to explain the mechanism of accumulation tides in the acid-soluble extracts were determined by high- of orotic acid and orotidine. Plausible explanations are pressure liquid chromatography. The concentrations of also offered for previously observed but unexplained find uracil and cytosine nucleotides decreased rapidly in the ings, i.e., the potentiation of the effects of ara-C by 6-aza- treated cells. Adenosine 5'-triphosphate and adenosine 5'- UR when 6-aza-UR is given prior to ara-C (2). diphosphate increased steadily, whereas guanosine 5'-di- phosphate decreased for a few hr before returning to the normal level. Orotic acid and orotidine, but no detectable MATERIALS AND METHODS amount of orotidine monophosphate (OMP), accumulated Chemicals. Fischer's medium, horse serum, and antibiot in treated cells. In homogenates of the cells harvested from ascitic fluids of mice, the presence of a membrane-bound ics necessary for the culture of L5178Y cells were obtained phosphatase with activity for OMP was demonstrated, from Grand Island Biological Co., Grand Island, N. -
Some Properties of Uridine-Cytidine Kinase from a Human Malignant Lymphoma1
[CANCER RESEARCH 39, 3102-3106, August 1979] 0008-5472/79/0039-0000$02.O0 Some Properties of Uridine-Cytidine Kinase from a Human Malignant Lymphoma1 Nahed K. Ahmed2and Arnold D. Welch Division of Biochemical and Clinical Pharmacology, St. Jude Children ‘sResearchHospital, Memphis, Tennessee38101 ABSTRACT partially purified from rat liver (4, 17), muninetumor cells (5, 6, 13), calf thymus (12), human neoplastic lymphoid cells (3), Unidine-cytidine kinase has been partially purified from a Ehrlich ascites tumor (21), Novikoff ascites rat tumor (17), and malignant human lymphoma, and some of its properties have also from Tetrahymena pyriformis (18). Urd-Cyd kinase cata been described. The apparent Michaelis constants for unidine lyzes the following reaction: and adenosine 5'-triphosphate are 7 x 1O@and 4 x i O@M, .. .. (Urd-Cyd)-kinase respectively. Maximal enzyme activity is observed between pH Urldine or cytldlne + NTP 6.5 and 8.5, and temperatures of incubation are observed uridine5'-monophosphateorcytidine5'-monophosphate+ NDP between 37 and 42°.The enzyme requires Mg2@for full enzy matic activity and exists in two isozymic forms, as indicated by where NTP is nucleoside tniphosphate, and NDP is nucleoside isoelectric focusing and column chromatography on Sepharose diphosphate. Previous partial purifications and the proper 6B. The roles of these two isozymes, i.e., the adult (I) and the ties of the enzyme have been reviewed (1, 2). The enzyme embryonic (II) forms, are not yet clear; conceivably, however, appears to require the nibofuranosyl moiety because 2'-deoxy they may have relevance to the problem of the development of nibonucleosides and certain arabinofuranosyl- and xylofura resistance to chemotherapeutic analogs that require unidine nosyl-containing nucleosides tested have not been phospho @ cytidine kinase for their ‘activation.― rylated (i 3, 18, 21). -

Eco-Evolutionary Feedbacks Drive Species Interactions
The ISME Journal (2014) 8, 1041–1054 & 2014 International Society for Microbial Ecology All rights reserved 1751-7362/14 www.nature.com/ismej ORIGINAL ARTICLE Eco-evolutionary feedbacks drive species interactions Andre´s Andrade-Domı´nguez1, Emmanuel Salazar1, Marı´a del Carmen Vargas-Lagunas1, Roberto Kolter2 and Sergio Encarnacio´n1 1Centro de Ciencias Geno´micas, Universidad Nacional Auto´noma de Me´xico, Cuernavaca, Me´xico and 2Department of Microbiology and Immunobiology, Harvard Medical School, Boston, MA, USA In the biosphere, many species live in close proximity and can thus interact in many different ways. Such interactions are dynamic and fall along a continuum between antagonism and cooperation. Because interspecies interactions are the key to understanding biological communities, it is important to know how species interactions arise and evolve. Here, we show that the feedback between ecological and evolutionary processes has a fundamental role in the emergence and dynamics of species interaction. Using a two-species artificial community, we demonstrate that ecological processes and rapid evolution interact to influence the dynamics of the symbiosis between a eukaryote (Saccharomyces cerevisiae) and a bacterium (Rhizobium etli). The simplicity of our experimental design enables an explicit statement of causality. The niche-constructing activities of the fungus were the key ecological process: it allowed the establishment of a commensal relationship that switched to ammensalism and provided the selective conditions necessary for the adaptive evolution of the bacteria. In this latter state, the bacterial population radiates into more than five genotypes that vary with respect to nutrient transport, metabolic strategies and global regulation. Evolutionary diversification of the bacterial populations has strong effects on the community; the nature of interaction subsequently switches from ammensalism to antagonism where bacteria promote yeast extinction. -

The Uricosuria and Orotic Aciduria Induced by 6- Azauridine
THE URICOSURIA AND OROTIC ACIDURIA INDUCED BY 6- AZAURIDINE Harold J. Fallon, … , Jerome Block, J. Edwin Seegmiller J Clin Invest. 1961;40(10):1906-1914. https://doi.org/10.1172/JCI104415. Research Article Find the latest version: https://jci.me/104415/pdf THE URICOSURIA AND OROTIC ACIDURIA INDUCED BY 6-AZAURIDINE * By HAROLD J. FALLON,t EMIL FREI, III, JEROME BLOCK AND J. EDWIN SEEGMILLER (From the laboratories of the Chemotherapy Service, General Medicine Branch of the National Cancer Institute and the Arthritis and Rheumatism Branch of the National Institute of Arthritis and Metabolic Diseases, Bethesda, Md.) (Submitted for publication May 8, 1961; accepted June 15, 1961) The pyrimidine analog 6-azauridine (6-AZUR) the National Cancer Institute, was occasionally interrupts the major pathway for the de novo bio- complicated by a pronounced crystalluria. The synthesis of pyrimidine nucleotides after its bio- crystals were separated into two components by logical conversion to 6-azauridine-5'-phosphate ion-exchange chromatography and were identified (6-AZUMP). 6-AZUMP is a competitive in- by their ultraviolet absorption spectra as uric and hibitor of the enzyme which decarboxylates oro- orotic acid. The appearance of orotidine in tissues tidylic acid (see Figure 1) (1, 2). 6-AZUR in- and orotic acid and orotidine in the urine of mice hibits bacterial reproduction as well as tumor receiving 6-AZUR has been previously noted (1, growth in animals (3-6). The latter effect has 3) and may be explained by the metabolic block recently led to its use in the experimental treat- produced by the drug. These compounds have ment of human leukemia and solid tumors (7, 8). -

Urinary Excretion of Orotic Acid, Orotidine and Other Pyrimidines in a Patient with Purine Nucleoside Phosphorylase Deficiency
419 Clinica Ckimica Acta, 93 (1979) 419-428 @ ElsevierlNorth-Holland Biomedical Press CCA 10221 URINARY EXCRETION OF OROTIC ACID, OROTIDINE AND OTHER PYRIMIDINES IN A PATIENT WITH PURINE NUCLEOSIDE PHOSPHORYLASE DEFICIENCY A.H. VAN GENNIP a, J. GRIFT a, P.K. DE BREE b, B.J.M. ZEGERS b, J.W. STOOP b and S.K. WADMAN b,* a Children’s Hospital, Het Emma Kinderziekenhuis, Spinozastraat 51, Amsterdam (The Netherlands) and b University Children’s Hospital, Het Wilhelmina Kinderziekenhuis, Nieuwe Gracht 137, Utrecht (The Netherlands) (Received December 8th, 1978) Summary Urinary orotidine and erotic acid have been determined in a patient with purine nucleoside phosphorylase (PNP) deficiency under various dietary and therapeutic conditions. For this purpose a new procedure for the analysis of both compounds has been developed, consisting of prefractionation with Dowex 1X8, followed by two HPLC steps on a ti Bondapak NH, and a /J Bon- dapak C,, column. With this method normal as well as slightly elevated excre- tions of erotic acid have been found in our patient. No evidence was obtained for inhibition of OPRT by purine (deoxy)nucleosides as a cause of pyrimidine starvation. A significant increase of urinary orotidine was found after loading with allopurinol. For comparison excretory values in a patient with omithine transcarbamylase deficiency and also in a patient with erotic aciduria type I are shown. The possible cause of the slight increase in urinary erotic acid in our patient has been discussed. Introduction Since in 1972 Giblett et al. [l] described the first case of a patient with severe combined immunodeficiency disease, lacking adenosine deaminase (ADA) activity in the erythrocytes, a number of similarly affected patients have been reported [Z--5]. -

Pyrimidine – Lecture
Pyrimidine – Lecture Raymond B. Birge, PhD Biochemistry & Molecular Biology Pyrimidine-Lecture Overview (i) Biosynthesis pathways (ii) Conversion of ribose bases to deoxyribose bases (iii) Chemotherapeutics, anti-metabolites Structure of Pyrimidines C= 2 oxy, 4 amino pyrimidine U= 2,4 dioxy pyrimidine T= 2,4 dioxy 5-methyl pyrmidine O= 2,4 dioxy 6 carboxy pyrimidine The nomenclature of purines and pyrimidines depends on their linkage to a pentose Cytosine Cytidine Cytidine Monophosphate Base Nucleoside* Nucleotide Base Base (P04 ester) * when the base is purine, then the nucleoside ends in OSINE (AdenOSINE, GuanOSINE, InOSINE) when the base is pyrimidine, then the nucleoside ends in IDINE (UrIDINE, CytIDINE, ThymIDINE) Pyrimidine Biosynthesis-I (occurs in cytosol) Pyrimidine biosynthesis begins with the assembly of the ring, then linked To ribose phosphate. Precursors are Glutamine (NH2), Bicarbonate (C) , and ATP (PO4). Q. Why is it advantageous to generate carbamoyl phosphate in the cytosol rather than the mitochondria? Pyrimidine Biosynthesis-II Carbamoyl phosphate synthase II, ATCase, and Dihydrooratase are linked in a single 240 kD polypeptide chain. The enzyme is sometimes referred to as CAD. Pyrimidine Metabolism-III Carbamyl Phosphate Aspartate Dihydrooratase Synthase II Transcarbamylase NH3 Carbamyl Carbamyl Glutamine HCO3 Phosphate Aspartate MgATP Dihydroorotic Aspartate H20 Acid 1. 2. 3. 4. Enzyme Cluster I DHOD Orotic Acid Uridine 5’ Orotidine PRPP Monophosphate 5’ Monophosphate (UMP) Orotate Orotidine 5’ Phosphate Phosphoribosyl Decarboxylase Transferase 6. 5. Enzyme Cluster II Pyrimidine Metabolism-III Carbamyl Phosphate Aspartate Dihydrooratase Synthase II Transcarbamylase CPS II ATCase DHO NH3 Carbamyl Carbamyl Glutamine HCO3 Phosphate Aspartate MgATP Dihydroorotic Aspartate H20 Acid 1. 2. 3. -

By Ctpsynthetase
Agric. Biol. Chem., 53 (1), 97- 102, 1989 97 Regulation of Pyrimidine Nucleotide Biosynthesis in Cytidine Deaminase-negative Mutants of Bacillus subtilisf Satoru ASAHi, Muneharu Doi, Yutaka TSUNEMI and Shun-ichi AKIYAMA Applied Microbiology Laboratories, Central Research Division, Takeda Chemical Industries, Ltd., Yodogawa-ku, Osaka 532, Japan Received July ll, 1988 The role of uridine and cytidine compoundsin regulating pyrimidine nucleotide biosynthesis de novo was studied with cytidine deaminase-negative mutants of Bacillus subtilis. In the wild type strain, the formation of six enzymes for uridine 5'-monophosphate (UMP)biosynthesis was severely repressed by exogenous cytidine or uracil, whereas the formation of the enzymesin a cytidine deaminase-negative mutant was repressed only by uracil. On the other hand, the formation of cytidine 5'-triphosphate (CTP) synthetase was not affected by uracil. This enzynme was repressed only when a cytidine deaminase-negative mutant was grown in the presence of excess cytidine. Studies on feedback inhibition also showed that the activity of CTP synthetase was inhibited by cytidine nucleotides, but not by uridine nucleotides. The synthesis de nove of uridine 5'-mono- distinguish between the effects of these two phosphate (UMP) in Bacillus subtilis is cata- compounds on gene expression, because they lyzed through the sequential actions of six are readily converted into each other through enzymes (Fig. 1). The expression of the pyr the sequential actions of several enzymes in- genes encoding these enzymesis repressed by volved in pyrimidine metabolism5) (Fig. 1). In adding uracil2) and appears to be coordinately the course of studies on the production of regulated.3) On the other hand, regulation of pyrimidine nucleosides by B. -

Biological and Clinical Aspects of Pyrimidine Metabolism
Pediat. Res. 8: 724-734 (1 974) A Review: Biological and Clinical Aspects of Pyrimidine Metabolism RODNEY L. LEVINE, NICHOLAS J. HOOGENRAAD, AND NORMAN KRETCHMER(' ) Division of Developmental Biology, Department of Pediatrics, Stanford University Medial Center, Stanford, California, USA Interest in pyrimidine nucleotide biosynthesis has stemmed or the amide nitrogen of glutamine), ATP, aspartate. and largely from the importance of these molecules as components ribose in the form of phosphoribosyl pyr~phos~hate.A of nucleic acids. Regulation of pyrimidine biosynthesis has reutilization ("salvage")-. mechanism is also available to cells. been most extensively investigated in micro-organisms in whereby preformed pyrimidine bases or nucleosides can be which cell division is effectively controlled by regulation of converted to the nucleotide by the addition of ribose the activity of the enzymes of the de novo pathway (77, 11 0). phosphate or phosphate. The relation between pyrimidine biosynthesis and cellular proliferation in mammals has become clearer with the DE NOVO MECHANISM accumulation of a considerable body of evidence which indicates that there is a close correlation between the activity De novo biosynthesis of uridylic acid required six enzymatic of enzymes involved in pyrimidine biosynthesis and the rate of steps (Fig. 1). In procaryotes one enzyme supplies the cellular proliferation. carbamyl phosphate required for both pyrimidine nucleotide The de novo pyrimidine biosynthetic pathway and the urea and arginine biosynthesis. Consequently, aspartate trans- cycle both utilize carbamyl phosphate, and important nitrogen carbamylase is the first enzyme unique to the de novo carrier. The biosynthesis of pyrimidine and purine nucleotides pyrimidine pathway and an important locus for control of the are connected by a common intermediate, phosphoribosyl pathway (77).