Orotic Aciduria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
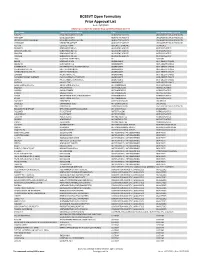
BCBSVT Open Formulary Prior Approval List
BCBSVT Open Formulary Prior Approval List As of: 10/27/2020 Helpful Tip: To search for a specific drug, use the find feature (Ctrl + F) Trade Name Chemical/Biological Name Class Prior Authorization Program FUSILEV LEVOLEUCOVORIN CALCIUM ADJUNCTIVE AGENTS UNCLASSIFIED DRUG PRODUCTS KHAPZORY LEVOLEUCOVORIN ADJUNCTIVE AGENTS UNCLASSIFIED DRUG PRODUCTS LEVOLEUCOVORIN CALCIUM LEVOLEUCOVORIN CALCIUM ADJUNCTIVE AGENTS UNCLASSIFIED DRUG PRODUCTS VISTOGARD URIDINE TRIACETATE ADJUNCTIVE AGENTS UNCLASSIFIED DRUG PRODUCTS ACTHAR CORTICOTROPIN ADRENAL HORMONES HORMONES BELRAPZO BENDAMUSTINE HCL ALKYLATING AGENTS ANTINEOPLASTICS BENDAMUSTINE HCL BENDAMUSTINE HCL ALKYLATING AGENTS ANTINEOPLASTICS BENDEKA BENDAMUSTINE HCL ALKYLATING AGENTS ANTINEOPLASTICS TREANDA BENDAMUSTINE HCL ALKYLATING AGENTS ANTINEOPLASTICS DAW (DISPENSE AS WRITTEN) ALL CUSTOM BELVIQ LORCASERIN HCL ANOREXIANTS ANTI‐OBESITY DRUGS BELVIQ XR LORCASERIN HCL ANOREXIANTS ANTI‐OBESITY DRUGS CONTRAVE ER NALTREXONE HCL/BUPROPION HCL ANOREXIANTS ANTI‐OBESITY DRUGS DIETHYLPROPION HCL DIETHYLPROPION HCL ANOREXIANTS ANTI‐OBESITY DRUGS DIETHYLPROPION HCL ER DIETHYLPROPION HCL ANOREXIANTS ANTI‐OBESITY DRUGS LOMAIRA PHENTERMINE HCL ANOREXIANTS ANTI‐OBESITY DRUGS PHENDIMETRAZINE TARTRATE PHENDIMETRAZINE TARTRATE ANOREXIANTS ANTI‐OBESITY DRUGS QSYMIA PHENTERMINE/TOPIRAMATE ANOREXIANTS ANTI‐OBESITY DRUGS SAXENDA LIRAGLUTIDE ANOREXIANTS ANTI‐OBESITY DRUGS ABIRATERONE ACETATE ABIRATERONE ACETATE ANTIANDROGENS ANTINEOPLASTICS ERLEADA APALUTAMIDE ANTIANDROGENS ANTINEOPLASTICS NUBEQA DAROLUTAMIDE ANTIANDROGENS -

Acute Toxicity of Cyanide in Aerobic Respiration: Theoretical and Experimental Support for Murburn Explanation
BioMol Concepts 2020; 11: 32–56 Research Article Open Access Kelath Murali Manoj*, Surjith Ramasamy, Abhinav Parashar, Daniel Andrew Gideon, Vidhu Soman, Vivian David Jacob, Kannan Pakshirajan Acute toxicity of cyanide in aerobic respiration: Theoretical and experimental support for murburn explanation https://doi.org/10.1515/bmc-2020-0004 received January 14, 2020; accepted February 19, 2020. of diffusible radicals and proton-equilibriums, explaining analogous outcomes in mOxPhos chemistry. Further, we Abstract: The inefficiency of cyanide/HCN (CN) binding demonstrate that superoxide (diffusible reactive oxygen with heme proteins (under physiological regimes) is species, DROS) enables in vitro ATP synthesis from demonstrated with an assessment of thermodynamics, ADP+phosphate, and show that this reaction is inhibited kinetics, and inhibition constants. The acute onset of by CN. Therefore, practically instantaneous CN ion-radical toxicity and CN’s mg/Kg LD50 (μM lethal concentration) interactions with DROS in matrix catalytically disrupt suggests that the classical hemeFe binding-based mOxPhos, explaining the acute lethal effect of CN. inhibition rationale is untenable to account for the toxicity of CN. In vitro mechanistic probing of Keywords: cyanide-poisoning; murburn concept; CN-mediated inhibition of hemeFe reductionist systems aerobic respiration; ATP-synthesis; hemoglobin; was explored as a murburn model for mitochondrial cytochrome oxidase; mitochondria; diffusible reactive oxidative phosphorylation (mOxPhos). The effect of CN oxygen species (DROS). in haloperoxidase catalyzed chlorine moiety transfer to small organics was considered as an analogous probe for phosphate group transfer in mOxPhos. Similarly, Introduction inclusion of CN in peroxidase-catalase mediated one- electron oxidation of small organics was used to explore Since the discovery of the dye Prussian blue (ferric electron transfer outcomes in mOxPhos, leading to water ferrocyanide, Fe7[CN]18) and the volatile prussic acid formation. -

35 Disorders of Purine and Pyrimidine Metabolism
35 Disorders of Purine and Pyrimidine Metabolism Georges van den Berghe, M.- Françoise Vincent, Sandrine Marie 35.1 Inborn Errors of Purine Metabolism – 435 35.1.1 Phosphoribosyl Pyrophosphate Synthetase Superactivity – 435 35.1.2 Adenylosuccinase Deficiency – 436 35.1.3 AICA-Ribosiduria – 437 35.1.4 Muscle AMP Deaminase Deficiency – 437 35.1.5 Adenosine Deaminase Deficiency – 438 35.1.6 Adenosine Deaminase Superactivity – 439 35.1.7 Purine Nucleoside Phosphorylase Deficiency – 440 35.1.8 Xanthine Oxidase Deficiency – 440 35.1.9 Hypoxanthine-Guanine Phosphoribosyltransferase Deficiency – 441 35.1.10 Adenine Phosphoribosyltransferase Deficiency – 442 35.1.11 Deoxyguanosine Kinase Deficiency – 442 35.2 Inborn Errors of Pyrimidine Metabolism – 445 35.2.1 UMP Synthase Deficiency (Hereditary Orotic Aciduria) – 445 35.2.2 Dihydropyrimidine Dehydrogenase Deficiency – 445 35.2.3 Dihydropyrimidinase Deficiency – 446 35.2.4 Ureidopropionase Deficiency – 446 35.2.5 Pyrimidine 5’-Nucleotidase Deficiency – 446 35.2.6 Cytosolic 5’-Nucleotidase Superactivity – 447 35.2.7 Thymidine Phosphorylase Deficiency – 447 35.2.8 Thymidine Kinase Deficiency – 447 References – 447 434 Chapter 35 · Disorders of Purine and Pyrimidine Metabolism Purine Metabolism Purine nucleotides are essential cellular constituents 4 The catabolic pathway starts from GMP, IMP and which intervene in energy transfer, metabolic regula- AMP, and produces uric acid, a poorly soluble tion, and synthesis of DNA and RNA. Purine metabo- compound, which tends to crystallize once its lism can be divided into three pathways: plasma concentration surpasses 6.5–7 mg/dl (0.38– 4 The biosynthetic pathway, often termed de novo, 0.47 mmol/l). starts with the formation of phosphoribosyl pyro- 4 The salvage pathway utilizes the purine bases, gua- phosphate (PRPP) and leads to the synthesis of nine, hypoxanthine and adenine, which are pro- inosine monophosphate (IMP). -

The Regulation of Carbamoyl Phosphate Synthetase-Aspartate Transcarbamoylase-Dihydroorotase (Cad) by Phosphorylation and Protein-Protein Interactions
THE REGULATION OF CARBAMOYL PHOSPHATE SYNTHETASE-ASPARTATE TRANSCARBAMOYLASE-DIHYDROOROTASE (CAD) BY PHOSPHORYLATION AND PROTEIN-PROTEIN INTERACTIONS Eric M. Wauson A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Pharmacology. Chapel Hill 2007 Approved by: Lee M. Graves, Ph.D. T. Kendall Harden, Ph.D. Gary L. Johnson, Ph.D. Aziz Sancar M.D., Ph.D. Beverly S. Mitchell, M.D. 2007 Eric M. Wauson ALL RIGHTS RESERVED ii ABSTRACT Eric M. Wauson: The Regulation of Carbamoyl Phosphate Synthetase-Aspartate Transcarbamoylase-Dihydroorotase (CAD) by Phosphorylation and Protein-Protein Interactions (Under the direction of Lee M. Graves, Ph.D.) Pyrimidines have many important roles in cellular physiology, as they are used in the formation of DNA, RNA, phospholipids, and pyrimidine sugars. The first rate- limiting step in the de novo pyrimidine synthesis pathway is catalyzed by the carbamoyl phosphate synthetase II (CPSase II) part of the multienzymatic complex Carbamoyl phosphate synthetase, Aspartate transcarbamoylase, Dihydroorotase (CAD). CAD gene induction is highly correlated to cell proliferation. Additionally, CAD is allosterically inhibited or activated by uridine triphosphate (UTP) or phosphoribosyl pyrophosphate (PRPP), respectively. The phosphorylation of CAD by PKA and ERK has been reported to modulate the response of CAD to allosteric modulators. While there has been much speculation on the identity of CAD phosphorylation sites, no definitive identification of in vivo CAD phosphorylation sites has been performed. Therefore, we sought to determine the specific CAD residues phosphorylated by ERK and PKA in intact cells. -

2018-2019 Targeted Medication Safety Best Practices for Hospitals
2018-2019 Targeted Medication Safety Best Practices for Hospitals The purpose of the Targeted Medication Safety Best Practices for Hospitals is to identify, inspire, and mobilize widespread, national adoption of consensus-based best practices for specific medication safety issues that continue to cause fatal and harmful errors in patients, despite repeated warnings in ISMP publications. Hospitals can focus their medication safety efforts over the next 2 years on these best practices, which are realistic and have been successfully adopted by numerous organizations. While targeted for the hospital-based setting, some best practices may be applicable to other healthcare settings. The Targeted Medication Safety Best Practices for Hospitals have been reviewed by an external expert advisory panel and approved by the ISMP Board of Trustees. Related issues of the ISMP Medication Safety Alert! are referenced after each best practice. ISMP encourages hospitals that have not implemented the 2016-2017 Targeted Medication Safety Best Practices for Hospitals to do so as a priority, while implementing the 2018-2019 best practices. Organizations need to focus on previous best practices 2, 3, 9 and 11 since these have the lowest implementation rate. Two of the 2016-2017 Targeted Medication Safety Best Practices for Hospitals (number 4 and 7) have been revised for 2018-2019. Best practices number 12 through 14 are new for 2018-2019. www.ismp.org BEST PRACTICE 1: Dispense vinCRIStine (and other vinca alkaloids) in a minibag of a compatible solution and not in a syringe. Rationale: Related ISMP Medication The goal of this best practice is to ensure that vinca alkaloids are Safety Alerts!: administered by the intravenous route only. -

Pharmacy and Poisons (Third and Fourth Schedule Amendment) Order 2017
Q UO N T FA R U T A F E BERMUDA PHARMACY AND POISONS (THIRD AND FOURTH SCHEDULE AMENDMENT) ORDER 2017 BR 111 / 2017 The Minister responsible for health, in exercise of the power conferred by section 48A(1) of the Pharmacy and Poisons Act 1979, makes the following Order: Citation 1 This Order may be cited as the Pharmacy and Poisons (Third and Fourth Schedule Amendment) Order 2017. Repeals and replaces the Third and Fourth Schedule of the Pharmacy and Poisons Act 1979 2 The Third and Fourth Schedules to the Pharmacy and Poisons Act 1979 are repealed and replaced with— “THIRD SCHEDULE (Sections 25(6); 27(1))) DRUGS OBTAINABLE ONLY ON PRESCRIPTION EXCEPT WHERE SPECIFIED IN THE FOURTH SCHEDULE (PART I AND PART II) Note: The following annotations used in this Schedule have the following meanings: md (maximum dose) i.e. the maximum quantity of the substance contained in the amount of a medicinal product which is recommended to be taken or administered at any one time. 1 PHARMACY AND POISONS (THIRD AND FOURTH SCHEDULE AMENDMENT) ORDER 2017 mdd (maximum daily dose) i.e. the maximum quantity of the substance that is contained in the amount of a medicinal product which is recommended to be taken or administered in any period of 24 hours. mg milligram ms (maximum strength) i.e. either or, if so specified, both of the following: (a) the maximum quantity of the substance by weight or volume that is contained in the dosage unit of a medicinal product; or (b) the maximum percentage of the substance contained in a medicinal product calculated in terms of w/w, w/v, v/w, or v/v, as appropriate. -

(12) Patent Application Publication (10) Pub. No.: US 2013/0059868 A1 Miner Et Al
US 2013 0059868A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2013/0059868 A1 Miner et al. (43) Pub. Date: Mar. 7, 2013 (54) TREATMENT OF GOUT Publication Classification (75) Inventors: Jeffrey Miner, San Diego, CA (US); Jean-Luc Girardet, San Diego, CA (51) Int. Cl. (US); Barry D. Quart, Encinitas, CA A613 L/496 (2006.01) (US) A6IP 29/00 (2006.01) (73) Assignee: Ardea Biociences, Inc., San Diego, CA A6IP 9/06 (2006.01) (US) A613 L/426 (2006.01) A 6LX3/59 (2006.01) (21) Appl. No.: 13/637,343 (52) U.S. Cl. ...................... 514/262.1: 514/384: 514/365 (22) PCT Fled: Mar. 29, 2011 (86) PCT NO.: PCT/US11A3O364 (57) ABSTRACT S371 (c)(1), (2), (4) Date: Oct. 24, 2012 Sodium 2-(5-bromo-4-(4-cyclopropyl-naphthalen-1-yl)-4H Related U.S. Application Data 1,2,4-triazol-3-ylthio)acetate is described. In addition, phar (60) Provisional application No. 61/319,014, filed on Mar. maceutical compositions and uses Such compositions for the 30, 2010. treatment of a variety of diseases and conditions. Patent Application Publication Mar. 7, 2013 Sheet 1 of 10 US 2013/00598.68A1 FIGURE 1 S. C SOC 55000 40 S. s 3. s Patent Application Publication Mar. 7, 2013 Sheet 2 of 10 US 2013/00598.68A1 ~~~::CC©>???>©><!--->?©><??--~~~~~·%~~}--~~~~~~~~*~~~~~~~~·;--~~~~~~~~~;~~~~~~~~~~}--~~~~~~~~*~~~~~~~~;·~~~~~ |×.> |||—||--~~~~ ¿*|¡ MSU No IL-1ra MSU50 IL-1ra MSU 100 IL-1ra MSU500 IL-1ra cells Only No IL-1 ra Patent Application Publication Mar. 7, 2013 Sheet 3 of 10 US 2013/00598.68A1 FIGURE 3 A: 50000 40000 R 30000 2 20000 10000 O -7 -6 -5 -4 -3 Lesinurad (log)M B: Lesinurad (log)M Patent Application Publication Mar. -

PHARMACEUTICAL APPENDIX to the TARIFF SCHEDULE 2 Table 1
Harmonized Tariff Schedule of the United States (2020) Revision 19 Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2020) Revision 19 Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names INN which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service CAS registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. -

Genome-Scale Fitness Profile of Caulobacter Crescentus Grown in Natural Freshwater
Supplemental Material Genome-scale fitness profile of Caulobacter crescentus grown in natural freshwater Kristy L. Hentchel, Leila M. Reyes Ruiz, Aretha Fiebig, Patrick D. Curtis, Maureen L. Coleman, Sean Crosson Tn5 and Tn-Himar: comparing gene essentiality and the effects of gene disruption on fitness across studies A previous analysis of a highly saturated Caulobacter Tn5 transposon library revealed a set of genes that are required for growth in complex PYE medium [1]; approximately 14% of genes in the genome were deemed essential. The total genome insertion coverage was lower in the Himar library described here than in the Tn5 dataset of Christen et al (2011), as Tn-Himar inserts specifically into TA dinucleotide sites (with 67% GC content, TA sites are relatively limited in the Caulobacter genome). Genes for which we failed to detect Tn-Himar insertions (Table S13) were largely consistent with essential genes reported by Christen et al [1], with exceptions likely due to differential coverage of Tn5 versus Tn-Himar mutagenesis and differences in metrics used to define essentiality. A comparison of the essential genes defined by Christen et al and by our Tn5-seq and Tn-Himar fitness studies is presented in Table S4. We have uncovered evidence for gene disruptions that both enhanced or reduced strain fitness in lake water and M2X relative to PYE. Such results are consistent for a number of genes across both the Tn5 and Tn-Himar datasets. Disruption of genes encoding three metabolic enzymes, a class C β-lactamase family protein (CCNA_00255), transaldolase (CCNA_03729), and methylcrotonyl-CoA carboxylase (CCNA_02250), enhanced Caulobacter fitness in Lake Michigan water relative to PYE using both Tn5 and Tn-Himar approaches (Table S7). -

Hereditary Orotic Aciduria and Megaloblastic Anaemia: a Second Case, with Response to Uridine
27 February 1965 Specialty Boards in U.S.A.-Buie MMDICAJRNL 547 particular instances by an additional specialist designated by to be completed more quickly; grading is more nearly accurate, the Review Committee. The reports and recommendations of and testing can be more extensive. As a rule the candidate these representatives are forwarded to the Review Committee, becomes eligible for his oral examination after he has passed along with other material prepared by the hospital, and the the " written." The oral examination varies with the require- evaluation by the Committee thus represents the joint action of ments and needs of various specialties. It is important because the Council, the Board, and in some cases the third national it is the last obstacle to be surmounted before the candidate is specialty organization. Depending upon the number of pro- certified. grammes in the various specialties, the related Review Com- Members of specialty boards are conscientious, honest, dedi- mittee meets from once to three times yearly for one or two cated persons who contribute freely of their time and effort, days. All residency programmes are resurveyed normally at often at great sacrifice, to advance the standards of their three-year intervals, but more frequently if the committee so specialty, without thought of personal aggrandizement or recom- directs. pense for their services. Like members of the faculty of a great The standards by which residency training, programmes are university sitting at a defence of a doctoral dissertation, they organized, and against which they are evaluated by the Review are imbued with a sense of their responsibility to determine the Committees, are published by the Council as the Essentials of competence of candidates who appear voluntarily before them Approved Residencies. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Wednesday, February 10, 2016 4 P.M
Wednesday, February 10, 2016 4 p.m. Oklahoma Health Care Authority 4345 N. Lincoln Blvd. Oklahoma City, OK 73105 The University of Oklahoma Health Sciences Center COLLEGE OF PHARMACY PHARMACY MANAGEMENT CONSULTANTS MEMORANDUM TO: Drug Utilization Review (DUR) Board Members FROM: Bethany Holderread, Pharm.D. SUBJECT: Packet Contents for DUR Board Meeting – February 10, 2016 DATE: February 1, 2016 Note: The DUR Board will meet at 4:00p.m. The meeting will be held at 4345 N Lincoln Blvd. Enclosed are the following items related to the February meeting. Material is arranged in order of the agenda. Call to Order Public Comment Forum Action Item – Approval of DUR Board Meeting Minutes – Appendix A Update on Medication Coverage Authorization Unit/Oral Viscous Lidocaine Claims Analysis Update – Appendix B Action Item – Vote to Prior Authorize Duopa™ (Carbidopa/Levodopa Enteral Suspension) and Rytary™ (Carbidopa/Levodopa Extended-Release Capsules) – Appendix C Action Item – Vote to Prior Authorize Cortisporin® and Pediotic® (Neomycin/Polymyxin B/Hydrocortisone Otic) – Appendix D Action Item – Vote to Prior Authorize Migranal® (Dihydroergotamine Nasal Spray) – Appendix E Action Item – Vote to Prior Authorize Strensiq™ (Asfotase Alfa) – Appendix F Action Item – Vote to Prior Authorize Varubi™ (Rolapitant) – Appendix G Action Item – Vote to Prior Authorize Xuriden™ (Uridine Triacetate) – Appendix H Annual Review of Gout Medications and 30-Day Notice to Prior Authorize Mitigare™ (Colchicine Capsules) and Zurampic® (Lesinurad) – Appendix I Annual Review of Seizure Medications and 30-Day Notice to Prior Authorize Spritam® (Levetiracetam) – Appendix J 30-Day Notice to Prior Authorize Solaraze® (Diclofenac Gel) – Appendix K Annual Review of Ulcerative Colitis Medications and 30-Day Notice to Prior Authorize Uceris® (Budesonide Extended-Release Tablets), Uceris® (Budesonide Rectal Foam), and Miscellaneous Mesalamine Products – Appendix L Annual Review of Ocular Allergy Medications and 30-Day Notice to Prior Authorize Pazeo® (Olopatadine Ophthalmic) – Appendix M ORI-4403 • P.O.