Current Topics in Gastritis - 2012
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

A Cura Di Antonia Cava 1 3 a Cura Di Antonia Cava 8 1
11381.1_cop 1381.1.13 17/02/20 16:36 Pagina 1 1 A cura di Antonia Cava 1 3 A cura di Antonia Cava 8 1 . IL GIOCO DEL KILLER 1 Questo volume raccoglie i contributi di studiosi provenienti da diversi ambiti disciplinari, A . Il gioco del killer accomunati dall’aver preso parte a un’iniziativa di formazione all’avanguardia ideata da C A Domenico Carzo: il Master “Esperto in intervento sociale minori e mafie”. I saggi proposti V analizzano le culture mafiose e la devianza minorile generando un serrato confronto tra dif - A Culture mafiose e minori ( ferenti prospettive teoriche e di ricerca. Una riflessione interdisciplinare che non solo inter - A preta il tema dei “figli di mafia”, ma esplora anche strategie che possano offrire un’alterna - C U tiva sociale, culturale e affettiva. R A D I Antonia Cava è professore associato di Sociologia dei processi culturali e comunicativi ) I presso il Dipartimento di Scienze Cognitive, Psicologiche, Pedagogiche e Studi Culturali L dell’Università degli Studi di Messina. Insegna Industria culturale e media studies e Sociologia G I della comunicazione e coordina il Master “Esperto in intervento sociale minori e mafie”. O Svolge attività di ricerca su pubblici televisivi, consumi culturali e immaginario mediale. Tra le C O sue pubblicazioni: # Foodpeople. Itinerari mediali e paesaggi gastronomici contemporanei D (Aracne 2018) e Noir Tv. La cronaca nera diventa format televisivo (FrancoAngeli 2013). E L K I L L E R SScienze de ll aCcomunicazione Collana diretta da Marino Livolsi Franc oAngeli ISBN 978-88-917-7285-5 -

Docenti Scuola Secondaria II Grado Elenco Provvisorio
Ministero dell’Istruzione, dell’Università e della Ricerca Ufficio Scolastico Regionale per il Lazio Ufficio VI - Ambito territoriale per la provincia di Roma GRADUATORIA PROVVISORIA DIRITTO ALLO STUDIO ANNO 2020 - SECONDO GRADO data prov. classe rapporto servizio titolo da anzianita' rinnovo fuori laurea ammesso n. nominativo nascita nascitaconcorso sede di servizio lavoro * part-time conseguire ** servizio permessi corso part-time con riserva 1 FARINA SILVIA 23/05/1964 RM A012 IPSEOA ARTUSI A NO A 11 SI NO NO NO 2 CARIATI GIANNI 04/10/1970 CS B016 IIS LUCA PACIOLO A NO A 10 SI NO NO SI 3 PELLE TERESA 04/09/1968 RM A046 I.I.S. CONFALONIERI DE CHIRICO A NO A 8 SI NO NO NO 4 PETRILLO RENATA 06/02/1968 NA A046 L.A. CARAVILLANI A SI A 6 SI NO NO NO 5 ORECCHINI CHIARA 31/01/1981 RM A054 IIS COPERNICO A NO A 4 SI NO NO NO 6 TRAPANNONE ALESSANDRA 17/05/1985 RM B011 I.T.A. EMILIO SERENI A NO A 2 SI NO NO NO 7 OLIVADESE SIMONA 08/11/1982 TO AB24 E. ROSSI B NO A 13 SI NO NO NO 8 IEMMA PIERA 20/04/1965 RC AA24 IIS L. LOMBARDO RADICE B NO A 9 SI NO NO NO 9 TARRA VINCENZA 28/07/1969 CE A046 I.I.S. DE AMICIS-CATTANEO B NO A 8 SI NO NO NO 10 BARBA MADDALENA 30/08/1989 SA B016 I.I.S. "ROSSELLINI" B NO A 7 SI NO NO NO 11 AGNELLO LUCA 14/02/1976 SR A020 I.S.I.S.S. -

Catálogo De Shortlatino ÍNDICE INDEX
European and Latin American short film market Catálogo de Shortlatino ÍNDICE INDEX Bienvenidos / Welcome .................................................... 3 Programa Shortlatino / Shortlatino program ............. 5 PAÍSES EUROPEOS / EUROPEAN COUNTRIES PAÍSES LATINOS / LATIN COUNTRIES Alemania / Germany ........................................................ 11 Argentina / Argentina .................................................. 299 Austria / Austria .............................................................. 40 Brasil / Brazil .................................................................. 303 Bélgica / Belgium ............................................................. 41 Chile / Chile ................................................................... 309 Bulgaria / Bulgaria ......................................................... 49 Colombia / Colombia ...................................................... 311 Dinamarca / Denmark .................................................... 50 Costa Rica / Costa Rica ................................................ 315 Eslovenia / Slovenia ........................................................ 51 Cuba / Cuba ..................................................................... 316 España / Spain ................................................................. 52 México / Mexico ............................................................... 317 Estonia / Estonia ........................................................... 168 Perú / Peru .................................................................... -

Langues, Accents, Prénoms & Noms De Famille
Les Secrets de la Septième Mer LLaanngguueess,, aacccceennttss,, pprréénnoommss && nnoommss ddee ffaammiillllee Il y a dans les Secrets de la Septième Mer une grande quantité de langues et encore plus d’accents. Paru dans divers supplément et sur le site d’AEG (pour les accents avaloniens), je vous les regroupe ici en une aide de jeu complète. D’ailleurs, à mon avis, il convient de les traiter à part des avantages, car ces langues peuvent être apprises après la création du personnage en dépensant des XP contrairement aux autres avantages. TTaabbllee ddeess mmaattiièèrreess Les différentes langues 3 Yilan-baraji 5 Les langues antiques 3 Les langues du Cathay 5 Théan 3 Han hua 5 Acragan 3 Khimal 5 Alto-Oguz 3 Koryo 6 Cymrique 3 Lanna 6 Haut Eisenör 3 Tashil 6 Teodoran 3 Tiakhar 6 Vieux Fidheli 3 Xian Bei 6 Les langues de Théah 4 Les langues de l’Archipel de Minuit 6 Avalonien 4 Erego 6 Castillian 4 Kanu 6 Eisenör 4 My’ar’pa 6 Montaginois 4 Taran 6 Ussuran 4 Urub 6 Vendelar 4 Les langues des autres continents 6 Vodacci 4 Les langages et codes secrets des différentes Les langues orphelines ussuranes 4 organisations de Théah 7 Fidheli 4 Alphabet des Croix Noires 7 Kosar 4 Assertions 7 Les langues de l’Empire du Croissant 5 Lieux 7 Aldiz-baraji 5 Heures 7 Atlar-baraji 5 Ponctuation et modificateurs 7 Jadur-baraji 5 Le code des pierres 7 Kurta-baraji 5 Le langage des paupières 7 Ruzgar-baraji 5 Le langage des “i“ 8 Tikaret-baraji 5 Le code de la Rose 8 Tikat-baraji 5 Le code 8 Tirala-baraji 5 Les Poignées de mains 8 1 Langues, accents, noms -

Estudio Sobre El Contexto Económico De Las Prácticas Audiovisuales Independientes En El Ámbito Artístico Catalán
Estudio sobre el contexto económico de las prácticas audiovisuales independientes en el ámbito artístico catalán: procesos de I+D y nuevos lechos de empleo Diciembre/2009 Estudio realizado por AAVC www.aavc.net (Associació d’Artistes Visuals de Catalunya) con el apoyo de la distribuidora de videoarte HAMACA www.hamacaonline.net y la coordinación de la productora cultural YProductions www.ypsite.net Índice 1. Introducción al objeto de estudio y metodología 3 1.1 Presentación del estudio 4 1.2 Metodología del estudio 6 1.2.1 Equipo investigador 6 1.2.2 Herramientas metodológicas 7 1.2.3 Marco temporal 7 1.2.4 Objeto de estudio 9 2. Aproximación a los perfiles y prácticas del sector 14 2.1 Videoarte 15 2.2 Documental independiente 29 2.3 Animación artística/experimental 38 2.4 Video creación en directo: Veejing y Live Cinema 49 2.5 Performance audiovisual y Videodanza 62 2.6 Cine experimental 70 2.7 Productoras audiovisuales independientes 81 2.8 Plataformas online del audiovisual independiente 94 3. Potencial de Innovación en el audiovisual independiente 113 3.1 Indicadores de Innovación Emergente en el Audiovisual Independiente 114 3.2 La transferencia de conocimiento 117 3.2.1 Áreas beneficiadas por la transferencia 117 3.2.2 Formas de transferencia 119 3.2.3 Conclusiones 123 3.3 Las redes en los procesos de I+D 124 3.3.1 Las redes como forma de trabajo 125 3.3.2 Estructura de las redes en los diferentes ámbitos 126 3.3.3 Redes sociales on-line 129 3.3.4 Conclusiones 129 3.4 La experimentación con nuevas tecnologías y modelos de software libre 131 3.4.1 Cultura Libre en el audiovisual independiente 132 3.4.2 Software libre en el audiovisual independiente 133 3.4.3 Viabilidad económica en proyectos de software libre 135 3.4.4 Conclusiones 137 3.5 Conclusiones 138 4. -
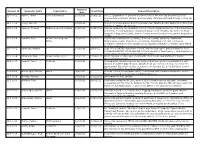
Requests Report
Received Request ID Requester Name Organization Closed Date Request Description Date 12-F-0001 Vahter, Tarmo Eesti Ajalehed AS 10/3/2011 3/19/2012 All U.S. Department of Defense documents about the meeting between Estonian president Arnold Ruutel (Ruutel) and Secretary of Defense Richard Cheney on July 19, 1991. 12-F-0002 Jeung, Michelle - 10/3/2011 - Copies of correspondence from Congresswoman Shelley Berkley and/or her office from January 1, 1999 to the present. 12-F-0003 Lemmer, Thomas McKenna Long & Aldridge 10/3/2011 11/22/2011 Records relating to the regulatory history of the following provisions of the Department of Defense Federal Acquisition Regulation Supplement (DFARS), the former Defense Acquisition Regulation (DAR), and the former Armed Services Procurement Regulation (ASPR). 12-F-0004 Tambini, Peter Weitz Luxenberg Law 10/3/2011 12/12/2011 Documents relating to the purchase, delivery, testing, sampling, installation, Office maintenance, repair, abatement, conversion, demolition, removal of asbestos containing materials and/or equipment incorporating asbestos-containing parts within its in the Pentagon. 12-F-0005 Ravnitzky, Michael - 10/3/2011 2/9/2012 Copy of the contract statement of work, and the final report and presentation from Contract MDA9720110005 awarded to the University of New Mexico. I would prefer to receive these documents electronically if possible. 12-F-0006 Claybrook, Rick Crowell & Moring LLP 10/3/2011 12/29/2011 All interagency or other agreements with effect to use USA Staffing for human resources management. 12-F-0007 Leopold, Jason Truthout 10/4/2011 - All documents revolving around the decision that was made to administer the anti- malarial drug MEFLOQUINE (aka LARIAM) to all war on terror detainees held at the Guantanamo Bay prison facility as stated in the January 23, 2002, Infection Control Standard Operating Procedure (SOP). -

Italian Films (Updated April 2011)
Language Laboratory Film Collection Wagner College: Campus Hall 202 Italian Films (updated April 2011): Agata and the Storm/ Agata e la tempesta (2004) Silvio Soldini. Italy The pleasant life of middle-aged Agata (Licia Maglietta) -- owner of the most popular bookstore in town -- is turned topsy-turvy when she begins an uncertain affair with a man 13 years her junior (Claudio Santamaria). Meanwhile, life is equally turbulent for her brother, Gustavo (Emilio Solfrizzi), who discovers he was adopted and sets off to find his biological brother (Giuseppe Battiston) -- a married traveling salesman with a roving eye. Bicycle Thieves/ Ladri di biciclette (1948) Vittorio De Sica. Italy Widely considered a landmark Italian film, Vittorio De Sica's tale of a man who relies on his bicycle to do his job during Rome's post-World War II depression earned a special Oscar for its devastating power. The same day Antonio (Lamberto Maggiorani) gets his vehicle back from the pawnshop, someone steals it, prompting him to search the city in vain with his young son, Bruno (Enzo Staiola). Increasingly, he confronts a looming desperation. Big Deal on Madonna Street/ I soliti ignoti (1958) Mario Monicelli. Italy Director Mario Monicelli delivers this deft satire of the classic caper film Rififi, introducing a bungling group of amateurs -- including an ex-jockey (Carlo Pisacane), a former boxer (Vittorio Gassman) and an out-of-work photographer (Marcello Mastroianni). The crew plans a seemingly simple heist with a retired burglar (Totó), who serves as a consultant. But this Italian job is doomed from the start. Blow up (1966) Michelangelo Antonioni. -

Sergo Kasvandik Dissertationes Medicinae Universitatis Tartuensis 303
SERGO KASVANDIK SERGO DISSERTATIONES MEDICINAE UNIVERSITATIS TARTUENSIS 303 The role of proteomic changes in proteomic endometrial of cells role – from theThe perspective fertility of and endometriosis SERGO K ASVA NDIK The role of proteomic changes in endometrial cells – from the perspective of fertility and endometriosis Tartu 2020 1 ISSN 1024-395X ISBN 978-9949-03-330-0 DISSERTATIONES MEDICINAE UNIVERSITATIS TARTUENSIS 303 1 DISSERTATIONES MEDICINAE UNIVERSITATIS TARTUENSIS 303 SERGO KASVANDIK The role of proteomic changes in endometrial cells – from the perspective of fertility and endometriosis 3 Institute of Clinical Medicine, University of Tartu, Estonia Dissertation is accepted for the commencement of the degree of Doctor of Philosophy in Medicine on the 15th of April, 2020 by the Council of the Faculty of Medicine, University of Tartu, Estonia. Supervisors: Andres Salumets, PhD, Professor of Reproductive Medicine, Department of Obstetrics and Gynaecology, Faculty of Medicine, Institute of Clinical Medicine, University of Tartu, Tartu, Estonia; Research Professor of Reproductive Genomics, Institute of Genomics, University of Tartu, Tartu, Estonia; Visiting Professor, Faculty of Medicine, University of Helsinki, Helsinki, Finland; Member of the Management Board, Competence Centre on Health Technologies AS, Tartu, Estonia. Maire Peters, PhD, Senior Research Fellow in Genetics, Department of Obstetrics and Gynaecology, Faculty of Medicine, Institute of Clinical Medicine, University of Tartu, Tartu, Estonia; Senior Researcher, Competence Centre on Health Technologies AS, Tartu, Estonia. Lauri Peil, PhD, Key Account and Technology Officer, Icosagen Cell Factory OÜ, Tartu, Estonia. Reviewers: Kalle Kilk, PhD, MD, Associate Professor of Medical Biochemistry / Senior Research Fellow in General Biochemistry, Department of Biochemistry, Institute of Biomedicine and Translational Medicine, Faculty of Medicine, University of Tartu, Tartu, Estonia. -

Foi Proposta Ho Conselho Forte Adoção Cruzeiro
¦ ; y-yíA: J4UTILAD0 | 'ff xiiara em sessões contínuas vot^M complemento ao Ato Adicional I m0"^^."^>if"*ft.<m.m*mm^m*i BRASIUA. 9 (Meridional) — O presidente do Conuolho, ar. T>n. credo Ncvea, cm despacho com o deputado Pinheiro Chagnn, lfder da Maioria da CAmura dos Depu. tados, foi cientificado haver n mencionado órgüo legislativo d* 1\ cldido, pela Mesa a pelo» lideres, realizar, n partir rfa próxima ter- ça_fe!ra, scasócs ininterruptas, Inclusive aos sábados, domingos • N.« 1.975 - CURITIBA, - . 55 u^ *¦¦ _____j * * SEXTA-FEIRA, 10 DE NOVEMBRO DE 1.961 ÓRGÃO DOS DIÁRIOS ASSOCIADOS - feriados, parn efeito de votar o ANO VII * projeto do lei complementar ao Ato ( Adicional e orçamento da "ífrtyy- i •y.y-.J ¦'-.:¦;¦ w-iíiV-. ••¦ ¦ æ"""--^—¦•'« Itcpúbllca. 'rr^l-__M__ii ¦~-AAAA"~'J_________C*«aJ___r'"'—"*+*%% •¦w$itf& Manifestações de %k$A'rA::-.? ¦***" */*.<+¦**>¦ VERUAS júbilo No despacho com o ministra da Via.im, o primeiro-ministro de '-*^B| terminou prioridades absoluta na na W\ >*•» f._____V ________iK____k____ *^T^3__________flS_ _________________!__________ _______ i de Moriroy como W. V Ammr1^ W%M _____Mr' mmmT*9*^rSmmT^^mmm^^mM___K BI BB liberação das verbas do DNOCS. posse referentes ar, regiões assoladas pelas secas: Bahia, Pernambuco, do Equador Sergipe e Alagoas. No mesmo presidente despacho, foi aprovado cm prln- C1U1TO, cfpio, o plano para unificação dos 11 (UPI — lUAItlO DO PAI'ANA') — O pi-aldent» Cnr- æ1>jt'4___E__r 1'AIIAKA ) — O pi'sidente Ç«irlo« loa Jiill0 An,,;.ni, im Monroy^decla- r*y>W mL ^i^^^^^tjMMflM ^_____H_____________ órgãos governamentais de vário» Jullo Araseinena Wpnroy fc_ una rou lio je que «*:u governo. -

Docenti Scuola Secondaria Di II Grado
Ministero dell’Istruzione, dell’Università e della Ricerca Ufficio Scolastico Regionale per il Lazio Ufficio VI - Ambito territoriale per la provincia di Roma GRADUATORIA DEFINITIVA DIRITTO ALLO STUDIO ANNO 2020 - SCUOLA SECONDARIA DI SECONDO GRADO data prov. classe rapporto servizio titolo da anzianita' rinnovo fuori laurea ammesso n. nominativo nascita nascitaconcorso sede di servizio lavoro * part-time conseguire ** servizio permessi corso part-time con riserva 1 LOVATO LUCA 14/01/1965 MN A018 LIEO CHRIS CAPPELL A NO A 26 SI NO NO NO 2 FARINA SILVIA 23/05/1964 RM A012 IPSEOA ARTUSI A NO A 11 SI NO NO NO 3 CARIATI GIANNI 04/10/1970 CS B016 IIS LUCA PACIOLO A NO A 10 SI NO NO NO 4 PATRONE TERESA 11/06/1969 NA A021 IIS LOI NETTUNO A NO A 9 SI NO NO NO 5 LEZZI CLELIA 10/11/1968 RM A054 LIEO CHRIS CAPPELL A NO A 8 SI NO NO NO 6 PELLE TERESA 04/09/1968 RM A046 I.I.S. CONFALONIERI DE CHIRICO A NO A 8 SI NO NO NO 7 PETRILLO RENATA 06/02/1968 NA A046 L.A. CARAVILLANI A SI A 6 SI NO NO NO 8 ORECCHINI CHIARA 31/01/1981 RM A054 IIS COPERNICO A NO A 4 SI NO NO NO 9 TRAPANNONE ALESSANDRA 17/05/1985 RM B011 I.T.A. EMILIO SERENI A NO A 2 SI NO NO NO 10 OLIVADESE SIMONA 08/11/1982 TO AB24 E. ROSSI B NO A 13 SI NO NO NO 11 IEMMA PIERA 20/04/1965 RC AA24 IIS L. -

European Union Film Festival WALTZ for MONICA (Sweden), March 8, 11
MAR 14 GAZETTE ■20 Vol. 42, No. 3 H H H H H See H Europe H H by Film H H H H 17th Annual European Union Film Festival WALTZ FOR MONICA (Sweden), March 8, 11 Complete schedule FREE SCHEDULE ■ NOT FOR SALE ■ For more information, on page 3 ALSO: CONVERSATIONS AT THE EDGE visit us online at: www.siskelfilmcenter.org $11 General Admission, $7 Students, $6 Members ■ To receive weekly updates and special offers, join our FOLLOW US! Join our email list email list at www.siskelfilmcenter.org at www.siskelfilmcenter.org H H H 17th Annual H H H H European Union H H H H H Film Festival CLOWNWISE (Czech Republic), March 9, 10 From March 7 through April 3, the Gene Siskel Film Center welcomes Guests include Austrian director Nicolas Neuhold with ANOTHER ONE you to the 17th Annual European Union Film Festival, the largest showcase OPENS, the first feature film with entirely improvised dialogue, on March 8 in North America for the cinema of the European Union nations. This and 13; Polish documentary director Anna Ferens with A PLACE TO STAND year’s festival presents the Chicago premieres of 64 new films from 26 EU on March 16; Chicago Reader film critic Ben Sachs discussing THE STUART nations, bringing the diverse national cultures of east and west Europe as HALL PROJECT on March 19; and German director David Sieveking close as your movie seat. presenting his very personal documentary FORGET ME NOT on March 21. Additional guests are pending as we go to press. -

Hospitals and Urbanism in Rome, 1200–1500 Brill’S Studies in Intellectual History
Hospitals and Urbanism in Rome, 1200–1500 Brill’s Studies in Intellectual History Series Editor Han van Ruler (Erasmus University Rotterdam) Editorial Board C.S. Celenza ( Johns Hopkins University, Baltimore) – M.L. Colish (Yale University) J.I. Israel (Institute for Advanced Study, Princeton) – A. Koba (University of Tokyo) M. Mugnai (Scuola Normale Superiore, Pisa) – W. Otten (University of Chicago) VOLUME 251 Brill’s Studies on Art, Art History, and Intellectual History General Editor Walter S. Melion (Emory University) VOLUME 12 The titles published in this series are listed at brill.com/bsai Hospitals and Urbanism in Rome, 1200–1500 By Carla Keyvanian LEIDEN | BOSTON Cover illustration: Remains of S. Urbano ai Pantani, the hospital of the Knights Hospitallers in Trajan’s Forum, Rome, early thirteenth century (photo © author). Library of Congress Cataloging-in-Publication Data Names: Keyvanian, Carla. Title: Hospitals and urbanism in Rome, 1200–1500 / by Carla Keyvanian. Description: Leiden : Brill, 2015. | Series: Brill’s studies in intellectual history, ISSN 0920-8607 ; volume 252 | Series: Brill’s studies on art, art history, and intellectual history ; volume 12 | Includes bibliographical references and index. Identifiers: LCCN 2015031751 | ISBN 9789004307544 (hardback : acid-free paper) | ISBN 9789004307551 (e-book) Subjects: LCSH: Hospital buildings—Italy—Rome—Design and construction—History—To 1500. | Public hospitals—Italy—Rome—History—To 1500. | Architecture and state—Italy—Rome—History—To 1500. | Urban development—Italy—Rome—History—To 1500. | City and town life—Italy—Rome—History— To 1500. | Rome (Italy)—Buildings, structures, etc. | Rome (Italy)—Social conditions. | Social control— Italy—Rome—History—To 1500. | Politics and culture—Italy—Rome—History—To 1500.