Functional Interpretation of ATAD3A Variants in Neuro-Mitochondrial Phenotypes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Chemical Modulation of Mitochondria–Endoplasmic Reticulum Contact Sites
cells Review Chemical Modulation of Mitochondria–Endoplasmic Reticulum Contact Sites 1 1, 1 1 Ana Paula Magalhães Rebelo , Federica Dal Bello y , Tomas Knedlik , Natasha Kaar , Fabio Volpin 1, Sang Hun Shin 1 and Marta Giacomello 1,2,* 1 Department of Biology, University of Padua, Via U. Bassi 58/B, 35121 Padua, Italy; [email protected] (A.P.M.R.); [email protected] (F.D.B.); [email protected] (T.K.); [email protected] (N.K.); [email protected] (F.V.); [email protected] (S.H.S.) 2 Department of Biomedical Sciences, University of Padua, Via U. Bassi 58/B, 35121 Padua, Italy * Correspondence: [email protected]; Tel.: +39-049-827-6300 Current Affiliation: Molecular Angiogenesis Laboratory, GIGA Research, University of Liège, B34, y Avenue de l’Hôpital, 1, 4000 Liège, Belgium. Received: 8 June 2020; Accepted: 2 July 2020; Published: 7 July 2020 Abstract: Contact sites between mitochondria and endoplasmic reticulum (ER) are points in which the two organelles are in close proximity. Due to their structural and functional complexity, their exploitation as pharmacological targets has never been considered so far. Notwithstanding, the number of compounds described to target proteins residing at these interfaces either directly or indirectly is rising. Here we provide original insight into mitochondria–ER contact sites (MERCs), with a comprehensive overview of the current MERCs pharmacology. Importantly, we discuss the considerable potential of MERCs to become a druggable target for the development of novel therapeutic strategies. Keywords: mitochondria–endoplasmic reticulum contact sites; mitochondria-associated membranes; pharmacology; drug targets; synthetic and biological compounds; neurodegeneration; diabetes; cancer 1. -

Identification of the Rs797045105 in the SERAC1 Gene by Whole-Exome Sequencing in a Patient Suspicious of MEGDEL Syndrome
Basic and Clinical July, August 2020, Volume 11, Number 4 News and Reports: Identification of the rs797045105 in the SERAC1 Gene by Whole-exome Sequencing in a Patient Suspicious of MEGDEL Syndrome Mina Zamani1,2 , Tahereh Seifi1,2, Jawaher Zeighami1, Neda Mazaheri1,2, Emad Jahangirnezhad1, Minoo Gholamzadeh1, Alireza Sed- aghat1,3* , Gholamreza Shariati1,4* , Hamid Galehdari1,2 1. Narges Medical Genetics and Prenatal Diagnosis Laboratory, Ahvaz, Iran. 2. Department of Genetics, Faculty of Science, Shahid Chamran University of Ahvaz, Ahvaz, Iran. 3. Diabetes Research Center, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran. 4. Department of Genetics, School of Medicine, Ahvaz Jundishapur University of medical Sciences, Ahvaz, Iran. Use your device to scan and read the article online Citation: Zamani, M., Seifi, T., Zeighami, J., Mazaheri, J., Mazaheri, N., & Jahangirnezhad, F., et al. (2020). Identification of the rs797045105 in the SERAC1 Gene by Whole-exome Sequencing in a Patient Suspicious of MEGDEL Syndrome. Basic and Clinical Neuroscience, 11(4), 549-556. http://dx.doi.org/10.32598/bcn.9.10.455 : http://dx.doi.org/10.32598/bcn.9.10.455 A B S T R A C T Introduction: Whole Exome Sequencing (WES) has been increasingly utilized in genetic determinants of various inherited diseases. Article info: Received: 13 Oct 2017 Methods: We applied WES for a patient presenting 3-Methylglutaconic Aciduria (MEG), First Revision:10 Nov 2017 Deafness (D), Encephalopathy (E), and Leigh-like (L) syndrome. Then Sanger sequencing was used for the detected variant validation. Accepted: 13 May 2019 Available Online: 01 Jul 2020 Results: We found an insertion, rs797045105 (chr6, 158571484, C>CCATG), in the SERAC1 gene with homozygous genotype in the patient and heterozygous genotype in her unaffected parents. -

Identification of Genetic Modifiers in Hereditary Spastic Paraplegias Due to SPAST/SPG4 Mutations Livia Parodi
Identification of genetic modifiers in Hereditary Spastic Paraplegias due to SPAST/SPG4 mutations Livia Parodi To cite this version: Livia Parodi. Identification of genetic modifiers in Hereditary Spastic Paraplegias due to SPAST/SPG4 mutations. Human health and pathology. Sorbonne Université, 2019. English. NNT : 2019SORUS317. tel-03141229 HAL Id: tel-03141229 https://tel.archives-ouvertes.fr/tel-03141229 Submitted on 15 Feb 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Sorbonne Université Institut du Cerveau et de la Moelle Épinière École Doctorale Cerveau-Cognition-Comportement Thèse de doctorat en Neurosciences Identification of genetic modifiers in Hereditary Spastic Paraplegias due to SPAST/SPG4 mutations Soutenue le 9 octobre 2019 par Livia Parodi Membres du jury : Pr Bruno Stankoff Président Pr Lesley Jones Rapporteur Dr Susanne de Bot Rapporteur Pr Christel Depienne Examinateur Pr Cyril Goizet Examinateur Pr Alexandra Durr Directeur de thèse Table of contents Abbreviations _________________________________________________________ -

Downregulation of Carnitine Acyl-Carnitine Translocase by Mirnas
Page 1 of 288 Diabetes 1 Downregulation of Carnitine acyl-carnitine translocase by miRNAs 132 and 212 amplifies glucose-stimulated insulin secretion Mufaddal S. Soni1, Mary E. Rabaglia1, Sushant Bhatnagar1, Jin Shang2, Olga Ilkayeva3, Randall Mynatt4, Yun-Ping Zhou2, Eric E. Schadt6, Nancy A.Thornberry2, Deborah M. Muoio5, Mark P. Keller1 and Alan D. Attie1 From the 1Department of Biochemistry, University of Wisconsin, Madison, Wisconsin; 2Department of Metabolic Disorders-Diabetes, Merck Research Laboratories, Rahway, New Jersey; 3Sarah W. Stedman Nutrition and Metabolism Center, Duke Institute of Molecular Physiology, 5Departments of Medicine and Pharmacology and Cancer Biology, Durham, North Carolina. 4Pennington Biomedical Research Center, Louisiana State University system, Baton Rouge, Louisiana; 6Institute for Genomics and Multiscale Biology, Mount Sinai School of Medicine, New York, New York. Corresponding author Alan D. Attie, 543A Biochemistry Addition, 433 Babcock Drive, Department of Biochemistry, University of Wisconsin-Madison, Madison, Wisconsin, (608) 262-1372 (Ph), (608) 263-9608 (fax), [email protected]. Running Title: Fatty acyl-carnitines enhance insulin secretion Abstract word count: 163 Main text Word count: 3960 Number of tables: 0 Number of figures: 5 Diabetes Publish Ahead of Print, published online June 26, 2014 Diabetes Page 2 of 288 2 ABSTRACT We previously demonstrated that micro-RNAs 132 and 212 are differentially upregulated in response to obesity in two mouse strains that differ in their susceptibility to obesity-induced diabetes. Here we show the overexpression of micro-RNAs 132 and 212 enhances insulin secretion (IS) in response to glucose and other secretagogues including non-fuel stimuli. We determined that carnitine acyl-carnitine translocase (CACT, Slc25a20) is a direct target of these miRNAs. -

Non Commercial Use Only
Trends in Evolutionary Biology 2017; volume 6:6514 Potential impact tions including insertional mutagenesis, of primate-specific SVA generation of deletions at the insertion site, Correspondence: Olga Vasieva, Institute of 3’ or 5’ transduction events, non-allelic Integrative Biology, Department of retrotransposons during homologous recombination and Comparative Genomics, University of the evolution of human exonisation.1-4 More than 10,000 TE inser- Liverpool, Liverpool, L69 7ZB, UK. cognitive function tions occurred in the human genome since Tel.: +44.151.795.4456 - Fax: +44.151.795.4406. human-chimpanzee divergence which were E-mail: [email protected] suggested to have implications on human 1 1 Key words: SVA, retrotransposons, hominoid, Olga Vasieva, Sultan Cetiner, evolution and especially its reproductive, 2 3 brain, evolution. Abigail Savage, Gerald G. Schumann, cognitive and immune functions that Vivien J. Bubb,2 John P. Quinn2 diverged strongly during a relative short Contributions: OV conceived of the study, 1Institute of Integrative Biology, time period.4,5 The large number of such designed and performed the systems analysis, University of Liverpool, UK; TEs in the genome makes an analysis of and drafted the manuscript, SC performed the 2Department of Molecular and Clinical their specific contribution to evolution very computational analysis and took part in draft- ing of the manuscript, AS has performed the Pharmacology, Institute of Translational difficult, and previous studies highlighted retrieval of SVA-associated gene -

SERAC1 Gene Serine Active Site Containing 1
SERAC1 gene serine active site containing 1 Normal Function The SERAC1 gene provides instructions for making a protein whose function is not completely understood. Studies suggest that the SERAC1 protein is involved in altering ( remodeling) certain fats called phospholipids, particularly a phospholipid called phosphatidylglycerol. Another phospholipid called cardiolipin is made from phosphatidylglycerol. Cardiolipin is a component of the membrane that surrounds cellular structures called mitochondria, which convert the energy from food into a form that cells can use, and is important for the proper functioning of these structures. Researchers believe that the SERAC1 protein is also involved in the movement of a waxy, fat-like substance called cholesterol within cells. Cholesterol is a structural component of cell membranes and plays a role in the production of certain hormones and digestive acids. It has important functions both before and after birth. Health Conditions Related to Genetic Changes MEGDEL syndrome At least 16 mutations in the SERAC1 gene have been found to cause MEGDEL syndrome. This condition is characterized by hearing loss, neurological problems, certain changes in the brain described as Leigh-like disease, and abnormally high amounts of an acid called 3-methylglutaconic acid in the urine. The SERAC1 gene mutations that cause this condition reduce the amount of SERAC1 protein that is produced or lead to production of a protein with little or no function. As a result, phosphatidylglycerol remodeling is impaired, which likely alters the composition of cardiolipin. Researchers speculate that the abnormal cardiolipin affects mitochondrial function, reducing cellular energy production and leading to the neurological and hearing problems characteristic of MEGDEL syndrome. -

Mutations and Protein Interaction Landscape Reveal Key Cellular Events Perturbed in Upper Motor Neurons with HSP and PLS
brain sciences Article Mutations and Protein Interaction Landscape Reveal Key Cellular Events Perturbed in Upper Motor Neurons with HSP and PLS Oge Gozutok 1, Benjamin Ryan Helmold 1 and P. Hande Ozdinler 1,2,3,4,* 1 Department of Neurology, Feinberg School of Medicine, Northwestern University, 303 E. Chicago Ave, Chicago, IL 60611, USA; [email protected] (O.G.); [email protected] (B.R.H.) 2 Center for Molecular Innovation and Drug Discovery, Center for Developmental Therapeutics, Chemistry of Life Processes Institute, Northwestern University, Evanston, IL 60611, USA 3 Mesulam Center for Cognitive Neurology and Alzheimer’s Disease, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA 4 Feinberg School of Medicine, Les Turner ALS Center at Northwestern University, Chicago, IL 60611, USA * Correspondence: [email protected]; Tel.: +1-(312)-503-2774 Abstract: Hereditary spastic paraplegia (HSP) and primary lateral sclerosis (PLS) are rare motor neuron diseases, which affect mostly the upper motor neurons (UMNs) in patients. The UMNs display early vulnerability and progressive degeneration, while other cortical neurons mostly remain functional. Identification of numerous mutations either directly linked or associated with HSP and PLS begins to reveal the genetic component of UMN diseases. Since each of these mutations are identified on genes that code for a protein, and because cellular functions mostly depend on protein- protein interactions, we hypothesized that the mutations detected in patients and the alterations in Citation: Gozutok, O.; Helmold, B.R.; protein interaction domains would hold the key to unravel the underlying causes of their vulnerability. Ozdinler, P.H. Mutations and Protein In an effort to bring a mechanistic insight, we utilized computational analyses to identify interaction Interaction Landscape Reveal Key Cellular Events Perturbed in Upper partners of proteins and developed the protein-protein interaction landscape with respect to HSP Motor Neurons with HSP and PLS. -

Downloaded from Ensembl
UCSF UC San Francisco Electronic Theses and Dissertations Title Detecting genetic similarity between complex human traits by exploring their common molecular mechanism Permalink https://escholarship.org/uc/item/1k40s443 Author Gu, Jialiang Publication Date 2019 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California by Submitted in partial satisfaction of the requirements for degree of in in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO AND UNIVERSITY OF CALIFORNIA, BERKELEY Approved: ______________________________________________________________________________ Chair ______________________________________________________________________________ ______________________________________________________________________________ ______________________________________________________________________________ ______________________________________________________________________________ Committee Members ii Acknowledgement This project would not have been possible without Prof. Dr. Hao Li, Dr. Jiashun Zheng and Dr. Chris Fuller at the University of California, San Francisco (UCSF) and Caribou Bioscience. The Li lab grew into a multi-facet research group consist of both experimentalists and computational biologists covering three research areas including cellular/molecular mechanism of ageing, genetic determinants of complex human traits and structure, function, evolution of gene regulatory network. Labs like these are the pillar of global success and reputation -

Hereditary Spastic Paraplegia Precision Panel Overview
Hereditary Spastic Paraplegia Precision Panel Overview Hereditary Spastic Paraplegia (HSP) includes a group of familial diseases that are characterized by progressive degeneration of the corticospinal tracts responsible for movement and sensation. HSPs are differentiated into “pure” forms if there is bladder involvement and “complicated” if there are additional neurologic or systemic abnormalities. The genetic classification of HSP is based upon the mode of inheritance, chromosomal locus, and causative mutation. Modes of inheritance include autosomal dominant, autosomal recessive and X-linked forms. The correlation of clinical classification with genetic classification is not yet defined as some genetic types of HSP are associated with both pure and complex phenotypes. The Igenomix Hereditary Spastic Paraplegia Precision Panel can serve as an accurate and directed diagnostic tool as well as a differential diagnosis of muscle weakness ultimately leading to a better management and prognosis of the disease. It provides a comprehensive analysis of the genes involved in this disease using next-generation sequencing (NGS) to fully understand the spectrum of relevant genes involved. Indications The Igenomix Hereditary Spastic Paraplegia Precision Panel is indicated in patients with a clinical suspicion or diagnosis presenting with or without the following manifestations: ‐ Leg weakness ‐ Leg spasticity ‐ Brisk tendon reflexes ‐ Extensor plantar reflex ‐ Bladder dysfunction (urinary urgency) ‐ Visual loss ‐ Hearing loss ‐ Difficulty speaking ‐ Peripheral neuropathy Clinical Utility The clinical utility of this panel is: - The genetic and molecular diagnosis for an accurate clinical diagnosis of a symptomatic patient. 1 - Early initiation of treatment with a multidisciplinary team for symptomatic treatment to improve mobility, increase range of motion and relieve discomfort using pharmacologic treatment as well as physical medicine and rehabilitation. -

Progressive Deafness–Dystonia Due to SERAC1 Mutations: a Study of 67 Cases
RESEARCH ARTICLE Progressive Deafness–Dystonia Due to SERAC1 Mutations: A Study of 67 Cases Roeltje R. Maas, MD,1 Katarzyna Iwanicka-Pronicka, MD,2 Sema Kalkan Ucar, MD,3 Bader Alhaddad, MD,4 Moeenaldeen AlSayed, MD,5,6 Mohammed A. Al-Owain, MD,5,6 Hamad I. Al-Zaidan, MD,5,6 Shanti Balasubramaniam, MBBS, FRACP.,7,8 Ivo Baric´, MD,9,10 Dalal K. Bubshait, MD,11 Alberto Burlina, MD,12 John Christodoulou, MD,13,14,15 Wendy K. Chung, MD,16 Roberto Colombo, MD,17,18 Niklas Darin, MD,19 Peter Freisinger, MD,20 Maria Teresa Garcia Silva, MD,21,22 Stephanie Grunewald, MD, PhD,23 Tobias B. Haack, MD,4,24 Peter M. van Hasselt, MD, PhD,25 Omar Hikmat, MD,26,27 Friederike Horster,€ MD,28 Pirjo Isohanni, MD,29,30 Khushnooda Ramzan, PhD,5,6 Reka Kovacs-Nagy, MD, PhD,4 Zita Krumina, MD, PhD,31 Elena Martin-Hernandez, MD,21,22 Johannes A. Mayr, PhD,32 Patricia McClean, MD,33 Linda De Meirleir, MD, PhD,34 Karin Naess, MD, PhD,35 Lock H. Ngu, MD,36 Magdalena Pajdowska, MD,37 Shamima Rahman, MA, PhD,38 Gillian Riordan, FCP,39 Lisa Riley, MD,14,15 Benjamin Roeben, MD,40,41 Frank Rutsch, MD,42 Rene Santer, MD,43 Manuel Schiff, MD, PhD,44 Martine Seders, PhD,45 Silvia Sequeira, MD,46 Wolfgang Sperl, MD, PhD,32 Christian Staufner, MD,28 Matthis Synofzik, MD,40,41 Robert W. Taylor, PhD,47 Joanna Trubicka, PhD,48 Konstantinos Tsiakas, MD,43 Ozlem Unal, MD,49 Evangeline Wassmer, MD,50 Yehani Wedatilake, MD,38 Toni Wolff, MD,51 Holger Prokisch, PhD,4,52 Eva Morava, MD, PhD,53 Ewa Pronicka, MD,54 Ron A. -
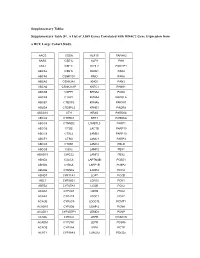
Supplementary Tables Supplementary Table S1. a List of 3,039 Genes
Supplementary Tables Supplementary Table S1. A List of 3,039 Genes Correlated with HDAC2 Gene Expression from a HCC Large Cohort Study. AACS CSDA KLF15 PAFAH2 AASS CSE1L KLF9 PAH ABAT CSE1L KLHL2 PAK1IP1 ABCA2 CSE1L KLKB1 PAK4 ABCA5 CSNK1G1 KMO PAK6 ABCA5 CSNK2A1 KNG1 PAN3 ABCA6 CSNK2A1P KNTC1 PANK1 ABCA9 CSPP1 KPNA2 PAOX ABCA9 CTBP2 KPNA4 PAPOLA ABCB1 CTDSP2 KPNA5 PAPSS1 ABCB4 CTDSPL2 KPNB1 PAQR8 ABCC10 CTH KRAS PARD6A ABCC6 CTHRC1 KRT3 PARD6G ABCC6 CTNND2 L3MBTL3 PARP1 ABCC6 CTSB LACTB PARP10 ABCC9 CTSL2 LAMB1 PARP10 ABCF1 CTSO LAMC1 PARP3 ABCG5 CTXN1 LAMC2 PBLD ABCG8 CUX2 LAMP2 PBX1 ABHD15 CWC22 LAMP2 PBX2 ABHD2 CXXC6 LAPTM4B PCBD1 ABHD5 CYB5A LARP1B PCBP2 ABHD6 CYB5D2 LARP4 PCCA ABHD7 CYP11A1 LCAT PCCB ABL1 CYP26B1 LCN12 PCK1 ABTB2 CYP27A1 LCOR PCK2 ACAA1 CYP2A7 LDHD PCK2 ACAA2 CYP2C8 LDOC1 PCK2 ACACB CYP2C9 LDOC1L PCMT1 ACAD10 CYP2D6 LEAP-2 PCNA ACAD11 CYP2D7P1 LEMD3 PCNP ACADL CYP2J2 LEPR PCSK1N ACADM CYP2W1 LEPR PCSK6 ACADS CYP3A4 LEPR PCTP ACAT1 CYP3A43 LGALS2 PDCD2 ACBD3 CYP3A5 LGALS7 PDCD2 ACBD4 CYP4A11 LIMA1 PDCL3 ACBD5 CYP4A22 LIMS2 PDE5A ACCN3 CYP4F11 LINGO1 PDK2 ACMSD CYP4F12 LIPG PDK4 ACO1 CYP4F2 LMAN1 PDS5B ACOT1 CYP4F3 LMAN2 PEA15 ACOT12 CYP7B1 LMAN2L PEBP1 ACOT4 CYP8B1 LMCD1 PECR ACOT8 DAK LMNB2 PELI1 ACOX1 DAO LOC120376 PELP1 ACOX2 DARS LOC123876 PEMT ACPL2 DBF4 LOC127295 PEPD ACSL1 DBN1 LOC130773 PER1 ACSM2A DBNDD2 LOC136143 PEX11G ACSM2B DBNL LOC143543 PEX13 ACSM2B DBT LOC143941 PEX14 ACSM2B DCAF16 LOC146439 PFDN5 ACSM3 DCAF7 LOC147710 PFKFB1 ACSM3 DCDC2 LOC158345 PGF ACSM5 DCTN2 LOC201725 PGM1 ACSM5 DCXR LOC220433