Supplementary Table S1. PCR Primer and Probe Set Information
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Acetyl Group Coordinated Progression Through the Catalytic Cycle of an Arylalkylamine N-Acetyltransferase
RESEARCH ARTICLE Acetyl group coordinated progression through the catalytic cycle of an arylalkylamine N-acetyltransferase Adam A. Aboalroub, Ashleigh B. Bachman, Ziming Zhang, Dimitra Keramisanou, David J. Merkler, Ioannis Gelis* Department of Chemistry, University of South Florida, Tampa, Florida, United States of America * [email protected] a1111111111 a1111111111 a1111111111 Abstract a1111111111 a1111111111 The transfer of an acetyl group from acetyl-CoA to an acceptor amine is a ubiquitous bio- chemical transformation catalyzed by Gcn5-related N-acetyltransferases (GNATs). Although it is established that the reaction proceeds through a sequential ordered mecha- nism, the role of the acetyl group in driving the ordered formation of binary and ternary com- OPEN ACCESS plexes remains elusive. Herein, we show that CoA and acetyl-CoA alter the conformation of the substrate binding site of an arylalkylamine N-acetyltransferase (AANAT) to facilitate Citation: Aboalroub AA, Bachman AB, Zhang Z, Keramisanou D, Merkler DJ, Gelis I (2017) Acetyl interaction with acceptor substrates. However, it is the presence of the acetyl group within group coordinated progression through the the catalytic funnel that triggers high affinity binding. Acetyl group occupancy is relayed catalytic cycle of an arylalkylamine N- through a conserved salt bridge between the P-loop and the acceptor binding site, and is acetyltransferase. PLoS ONE 12(5): e0177270. manifested as differential dynamics in the CoA and acetyl-CoA-bound states. The capacity https://doi.org/10.1371/journal.pone.0177270 of the acetyl group carried by an acceptor to promote its tight binding even in the absence of Editor: Viswanathan V. Krishnan, California State CoA, but also its mutually exclusive position to the acetyl group of acetyl-CoA underscore its University Fresno, UNITED STATES importance in coordinating the progression of the catalytic cycle. -

Supplemental Table S1
Entrez Gene Symbol Gene Name Affymetrix EST Glomchip SAGE Stanford Literature HPA confirmed Gene ID Profiling profiling Profiling Profiling array profiling confirmed 1 2 A2M alpha-2-macroglobulin 0 0 0 1 0 2 10347 ABCA7 ATP-binding cassette, sub-family A (ABC1), member 7 1 0 0 0 0 3 10350 ABCA9 ATP-binding cassette, sub-family A (ABC1), member 9 1 0 0 0 0 4 10057 ABCC5 ATP-binding cassette, sub-family C (CFTR/MRP), member 5 1 0 0 0 0 5 10060 ABCC9 ATP-binding cassette, sub-family C (CFTR/MRP), member 9 1 0 0 0 0 6 79575 ABHD8 abhydrolase domain containing 8 1 0 0 0 0 7 51225 ABI3 ABI gene family, member 3 1 0 1 0 0 8 29 ABR active BCR-related gene 1 0 0 0 0 9 25841 ABTB2 ankyrin repeat and BTB (POZ) domain containing 2 1 0 1 0 0 10 30 ACAA1 acetyl-Coenzyme A acyltransferase 1 (peroxisomal 3-oxoacyl-Coenzyme A thiol 0 1 0 0 0 11 43 ACHE acetylcholinesterase (Yt blood group) 1 0 0 0 0 12 58 ACTA1 actin, alpha 1, skeletal muscle 0 1 0 0 0 13 60 ACTB actin, beta 01000 1 14 71 ACTG1 actin, gamma 1 0 1 0 0 0 15 81 ACTN4 actinin, alpha 4 0 0 1 1 1 10700177 16 10096 ACTR3 ARP3 actin-related protein 3 homolog (yeast) 0 1 0 0 0 17 94 ACVRL1 activin A receptor type II-like 1 1 0 1 0 0 18 8038 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 1 0 0 0 0 19 8751 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 1 0 0 0 0 20 8728 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 1 0 0 0 0 21 81792 ADAMTS12 ADAM metallopeptidase with thrombospondin type 1 motif, 12 1 0 0 0 0 22 9507 ADAMTS4 ADAM metallopeptidase with thrombospondin type 1 -

Identification of Key Candidate Genes and Molecular Pathways in White Fat
Pan et al. Human Genomics (2019) 13:55 https://doi.org/10.1186/s40246-019-0239-x PRIMARY RESEARCH Open Access Identification of key candidate genes and molecular pathways in white fat browning: an anti-obesity drug discovery based on computational biology Yuyan Pan, Jiaqi Liu* and Fazhi Qi* Abstract Background: Obesity—with its increased risk of obesity-associated metabolic diseases—has become one of the greatest public health epidemics of the twenty-first century in affluent countries. To date, there are no ideal drugs for treating obesity. Studies have shown that activation of brown adipose tissue (BAT) can promote energy consumption and inhibit obesity, which makes browning of white adipose tissue (WAT) a potential therapeutic target for obesity. Our objective was to identify genes and molecular pathways associated with WAT and the activation of BAT to WAT browning, by using publicly available data and computational tools; this knowledge might help in targeting relevant signaling pathways for treating obesity and other related metabolic diseases. Results: In this study, we used text mining to find out genes related to brown fat and white fat browning. Combined with biological process and pathway analysis in GeneCodis and protein-protein interaction analysis by using STRING and Cytoscape, a list of high priority target genes was developed. The Human Protein Atlas was used to analyze protein expression. Candidate drugs were derived on the basis of the drug-gene interaction analysis of the final genes. Our study identified 18 genes representing 6 different pathways, targetable by a total of 33 drugs as possible drug treatments. The final list included 18 peroxisome proliferator-activated receptor gamma (PPAR-γ) agonists, 4 beta 3 adrenoceptor (β3-AR) agonists, 1 insulin sensitizer, 3 insulins, 6 lipase clearing factor stimulants and other drugs. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

A Clinicopathological and Molecular Genetic Analysis of Low-Grade Glioma in Adults
A CLINICOPATHOLOGICAL AND MOLECULAR GENETIC ANALYSIS OF LOW-GRADE GLIOMA IN ADULTS Presented by ANUSHREE SINGH MSc A thesis submitted in partial fulfilment of the requirements of the University of Wolverhampton for the degree of Doctor of Philosophy Brain Tumour Research Centre Research Institute in Healthcare Sciences Faculty of Science and Engineering University of Wolverhampton November 2014 i DECLARATION This work or any part thereof has not previously been presented in any form to the University or to any other body whether for the purposes of assessment, publication or for any other purpose (unless otherwise indicated). Save for any express acknowledgments, references and/or bibliographies cited in the work, I confirm that the intellectual content of the work is the result of my own efforts and of no other person. The right of Anushree Singh to be identified as author of this work is asserted in accordance with ss.77 and 78 of the Copyright, Designs and Patents Act 1988. At this date copyright is owned by the author. Signature: Anushree Date: 30th November 2014 ii ABSTRACT The aim of the study was to identify molecular markers that can determine progression of low grade glioma. This was done using various approaches such as IDH1 and IDH2 mutation analysis, MGMT methylation analysis, copy number analysis using array comparative genomic hybridisation and identification of differentially expressed miRNAs using miRNA microarray analysis. IDH1 mutation was present at a frequency of 71% in low grade glioma and was identified as an independent marker for improved OS in a multivariate analysis, which confirms the previous findings in low grade glioma studies. -

Methods in and Applications of the Sequencing of Short Non-Coding Rnas" (2013)
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2013 Methods in and Applications of the Sequencing of Short Non- Coding RNAs Paul Ryvkin University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, Genetics Commons, and the Molecular Biology Commons Recommended Citation Ryvkin, Paul, "Methods in and Applications of the Sequencing of Short Non-Coding RNAs" (2013). Publicly Accessible Penn Dissertations. 922. https://repository.upenn.edu/edissertations/922 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/922 For more information, please contact [email protected]. Methods in and Applications of the Sequencing of Short Non-Coding RNAs Abstract Short non-coding RNAs are important for all domains of life. With the advent of modern molecular biology their applicability to medicine has become apparent in settings ranging from diagonistic biomarkers to therapeutics and fields angingr from oncology to neurology. In addition, a critical, recent technological development is high-throughput sequencing of nucleic acids. The convergence of modern biotechnology with developments in RNA biology presents opportunities in both basic research and medical settings. Here I present two novel methods for leveraging high-throughput sequencing in the study of short non- coding RNAs, as well as a study in which they are applied to Alzheimer's Disease (AD). The computational methods presented here include High-throughput Annotation of Modified Ribonucleotides (HAMR), which enables researchers to detect post-transcriptional covalent modifications ot RNAs in a high-throughput manner. In addition, I describe Classification of RNAs by Analysis of Length (CoRAL), a computational method that allows researchers to characterize the pathways responsible for short non-coding RNA biogenesis. -

A Comprehensive Network and Pathway Analysis of Human Deafness Genes
Otology & Neurotology 34:961Y970 Ó 2013, Otology & Neurotology, Inc. A Comprehensive Network and Pathway Analysis of Human Deafness Genes *Georgios A. Stamatiou and †Konstantina M. Stankovic *Department of Otolaryngology, Hippokration General Hospital, University of Athens, Athens, Greece; and ÞDepartment of Otology and Laryngology, Harvard Medical School and Department of Otolaryngology, Massachusetts Eye and Ear Infirmary, Boston, Massachusetts, U.S.A. Objective: To perform comprehensive network and pathway factor beta1 (TGFB1) for Group 1, MAPK3/MAPK1 MAP kinase analyses of the genes known to cause genetic hearing loss. (ERK 1/2) and the G protein coupled receptors (GPCR) for Study Design: In silico analysis of deafness genes using inge- Group 2, and TGFB1 and hepatocyte nuclear factor 4 alpha (HNF4A) nuity pathway analysis (IPA). for Group 3. The nodal molecules included not only those known Methods: Genes relevant for hearing and deafness were iden- to be associated with deafness (GPCR), or with predisposition to tified through PubMed literature searches and the Hereditary otosclerosis (TGFB1), but also novel genes that have not been Hearing Loss Homepage. The genes were assembled into 3 groups: described in the cochlea (HNF4A) and signaling kinases (ERK 1/2). 63 genes that cause nonsyndromic deafness, 107 genes that cause Conclusion: A number of molecules that are likely to be key nonsyndromic or syndromic sensorineural deafness, and 112 genes mediators of genetic hearing loss were identified through three associated with otic capsule development and malformations. Each different network and pathway analyses. The molecules included group of genes was analyzed using IPA to discover the most new candidate genes for deafness. -

Comparative Biochemistry and Physiology, Part D, Vol. 5, Pp. 45-54 (2010)
Comparative genomics and proteomics of vertebrate diacylglycerol acyltransferase (DGAT), acyl CoA wax alcohol acyltransferase (AWAT) and monoacylglycerol acyltransferase (MGAT) Author Holmes, Roger S Published 2010 Journal Title Comparative Biochemistry and Physiology, Part D DOI https://doi.org/10.1016/j.cbd.2009.09.004 Copyright Statement © 2010 Elsevier. This is the author-manuscript version of this paper. Reproduced in accordance with the copyright policy of the publisher. Please refer to the journal's website for access to the definitive, published version. Downloaded from http://hdl.handle.net/10072/36786 Griffith Research Online https://research-repository.griffith.edu.au Comparative Biochemistry and Physiology, Part D, Vol. 5, pp. 45-54 (2010) COMPARATIVE GENOMICS AND PROTEOMICS OF VERTEBRATE DIACYLGLYCEROL ACYLTRANSFERASE (DGAT), ACYL CoA WAX ALCOHOL ACYLTRANSFERASE (AWAT) AND MONOACYLGLYCEROL ACYLTRANSFERASE (MGAT) Roger S Holmes School of Biomolecular and Physical Sciences, Griffith University, Nathan 4111 Brisbane Queensland Australia Email: [email protected] Keywords: Diacylglycerol acyltransferase-Monoacylglycerol transferase-Human- Mouse-Opossum-Zebrafish-Genetics-Evolution-X chromosome Running Head: Genomics and proteomics of vertebrate acylglycerol acyltransferases ABSTRACT BLAT (BLAST-Like Alignment Tool) analyses of the opossum (Monodelphis domestica) and zebrafish (Danio rerio) genomes were undertaken using amino acid sequences of the acylglycerol acyltransferase (AGAT) superfamily. Evidence is reported for 8 opossum monoacylglycerol acyltransferase-like (MGAT) (E.C. 2.3.1.22) and diacylglycerol acyltransferase-like (DGAT) (E.C. 2.3.1.20) genes and proteins, including DGAT1, DGAT2, DGAT2L6 (DGAT2-like protein 6), AWAT1 (acyl-CoA wax alcohol acyltransferase 1), AWAT2, MGAT1, MGAT2 and MGAT3. Three of these genes (AWAT1, AWAT2 and DGAT2L6) are closely localized on the opossum X chromosome. -
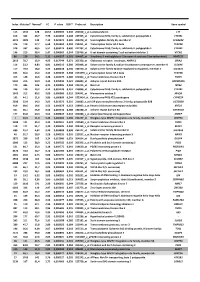
Index Mutated* Normal* FC P Value FDR** Probe Set Description Gene Symbol
Index Mutated* Normal* FC P value FDR** Probe set Description Gene symbol 113 1342 128 10,52 0,000503 0,240 202018_s_at Lactotransferrin LTF 616 362 46,7 7,76 0,003493 0,308 237395_at Cytochrome P450, family 4, subfamily Z, polypeptide 1 CYP4Z1 3073 1009 142 7,10 0,021674 0,385 206378_at Secretoglobin, family 2A, member 2 SCGB2A2 376 119 17,7 6,68 0,001962 0,284 214451_at Transcription factor AP-2 beta TFAP2B 578 337 60,5 5,57 0,003174 0,300 227702_at Cytochrome P450, family 4, subfamily X, polypeptide 1 CYP4X1 146 210 38,4 5,47 0,000669 0,244 219768_at V-set domain containing T cell activation inhibitor 1 VTCN1 86 129 24,2 5,31 0,000327 0,201 204607_at 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 (mitochondrial) HMGCS2 2613 78,7 15,9 4,95 0,017744 0,371 205358_at Glutamate receptor, ionotropic, AMPA 2 GRIA2 116 23,2 4,83 4,81 0,000513 0,240 203908_at Solute carrier family 4, sodium bicarbonate cotransporter, member 4 SLC4A4 117 79,4 18,0 4,42 0,000518 0,240 239723_at Solute carrier family 40 (iron-regulated transporter), member 1 SLC40A1 625 56,6 13,0 4,34 0,003549 0,308 1553394_a_atTranscription factor AP-2 beta TFAP2B 413 148 34,6 4,28 0,002175 0,286 240304_s_at Transmembrane channel-like 5 TMC5 5181 216 50,9 4,25 0,039946 0,421 223864_at Ankyrin repeat domain 30A ANKRD30A 179 446 106 4,21 0,000826 0,244 223315_at Netrin 4 NTN4 246 120 29,2 4,12 0,001128 0,251 210096_at Cytochrome P450, family 4, subfamily B, polypeptide 1 CYP4B1 1043 312 80,2 3,89 0,006080 0,319 204041_at Monoamine oxidase B MAOB 192 44,1 11,6 3,80 0,000899 0,244 -

Structural Capacitance in Protein Evolution and Human Diseases
Structural Capacitance in Protein Evolution and Human Diseases Adrian Woolfson, Ashley Buckle, Natalie Borg, Geoffrey Webb, Itamar Kass, Malcolm Buckle, Jiangning Song, Chen Li, Liah Clark, Rory Zhang, et al. To cite this version: Adrian Woolfson, Ashley Buckle, Natalie Borg, Geoffrey Webb, Itamar Kass, et al.. Structural Ca- pacitance in Protein Evolution and Human Diseases. Journal of Molecular Biology, Elsevier, 2018, 10.1016/j.jmb.2018.06.051. hal-02368321 HAL Id: hal-02368321 https://hal.archives-ouvertes.fr/hal-02368321 Submitted on 20 Nov 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Structural Capacitance in Protein Evolution and Human Diseases Chen Li, Liah Clark, Rory Zhang, Benjamin Porebski, Julia Mccoey, Natalie Borg, Geoffrey Webb, Itamar Kass, Malcolm Buckle, Jiangning Song, etal. To cite this version: Chen Li, Liah Clark, Rory Zhang, Benjamin Porebski, Julia Mccoey, et al.. Structural Capaci- tance in Protein Evolution and Human Diseases. Journal of Molecular Biology, Elsevier, 2018, 10.1016/j.jmb.2018.06.051. hal-02368321 HAL Id: hal-02368321 https://hal.archives-ouvertes.fr/hal-02368321 Submitted on 20 Nov 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. -

ITRAQ-Based Quantitative Proteomic Analysis of Processed Euphorbia Lathyris L
Zhang et al. Proteome Science (2018) 16:8 https://doi.org/10.1186/s12953-018-0136-6 RESEARCH Open Access ITRAQ-based quantitative proteomic analysis of processed Euphorbia lathyris L. for reducing the intestinal toxicity Yu Zhang1, Yingzi Wang1*, Shaojing Li2*, Xiuting Zhang1, Wenhua Li1, Shengxiu Luo1, Zhenyang Sun1 and Ruijie Nie1 Abstract Background: Euphorbia lathyris L., a Traditional Chinese medicine (TCM), is commonly used for the treatment of hydropsy, ascites, constipation, amenorrhea, and scabies. Semen Euphorbiae Pulveratum, which is another type of Euphorbia lathyris that is commonly used in TCM practice and is obtained by removing the oil from the seed that is called paozhi, has been known to ease diarrhea. Whereas, the mechanisms of reducing intestinal toxicity have not been clearly investigated yet. Methods: In this study, the isobaric tags for relative and absolute quantitation (iTRAQ) in combination with the liquid chromatography-tandem mass spectrometry (LC-MS/MS) proteomic method was applied to investigate the effects of Euphorbia lathyris L. on the protein expression involved in intestinal metabolism, in order to illustrate the potential attenuated mechanism of Euphorbia lathyris L. processing. Differentially expressed proteins (DEPs) in the intestine after treated with Semen Euphorbiae (SE), Semen Euphorbiae Pulveratum (SEP) and Euphorbiae Factor 1 (EFL1) were identified. The bioinformatics analysis including GO analysis, pathway analysis, and network analysis were done to analyze the key metabolic pathways underlying the attenuation mechanism through protein network in diarrhea. Western blot were performed to validate selected protein and the related pathways. Results: A number of differentially expressed proteins that may be associated with intestinal inflammation were identified. -

Transcriptomic and Proteomic Profiling Provides Insight Into
BASIC RESEARCH www.jasn.org Transcriptomic and Proteomic Profiling Provides Insight into Mesangial Cell Function in IgA Nephropathy † † ‡ Peidi Liu,* Emelie Lassén,* Viji Nair, Celine C. Berthier, Miyuki Suguro, Carina Sihlbom,§ † | † Matthias Kretzler, Christer Betsholtz, ¶ Börje Haraldsson,* Wenjun Ju, Kerstin Ebefors,* and Jenny Nyström* *Department of Physiology, Institute of Neuroscience and Physiology, §Proteomics Core Facility at University of Gothenburg, University of Gothenburg, Gothenburg, Sweden; †Division of Nephrology, Department of Internal Medicine and Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan; ‡Division of Molecular Medicine, Aichi Cancer Center Research Institute, Nagoya, Japan; |Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden; and ¶Integrated Cardio Metabolic Centre, Karolinska Institutet Novum, Huddinge, Sweden ABSTRACT IgA nephropathy (IgAN), the most common GN worldwide, is characterized by circulating galactose-deficient IgA (gd-IgA) that forms immune complexes. The immune complexes are deposited in the glomerular mesangium, leading to inflammation and loss of renal function, but the complete pathophysiology of the disease is not understood. Using an integrated global transcriptomic and proteomic profiling approach, we investigated the role of the mesangium in the onset and progression of IgAN. Global gene expression was investigated by microarray analysis of the glomerular compartment of renal biopsy specimens from patients with IgAN (n=19) and controls (n=22). Using curated glomerular cell type–specific genes from the published literature, we found differential expression of a much higher percentage of mesangial cell–positive standard genes than podocyte-positive standard genes in IgAN. Principal coordinate analysis of expression data revealed clear separation of patient and control samples on the basis of mesangial but not podocyte cell–positive standard genes.