Whole-Exome Analysis Reveals X-Linked Recessive Mutations Underlying Premature Ovarian Failure Using Galaxy Platform Abstract A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Snps) Distant from Xenobiotic Response Elements Can Modulate Aryl Hydrocarbon Receptor Function: SNP-Dependent CYP1A1 Induction S
Supplemental material to this article can be found at: http://dmd.aspetjournals.org/content/suppl/2018/07/06/dmd.118.082164.DC1 1521-009X/46/9/1372–1381$35.00 https://doi.org/10.1124/dmd.118.082164 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 46:1372–1381, September 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Single Nucleotide Polymorphisms (SNPs) Distant from Xenobiotic Response Elements Can Modulate Aryl Hydrocarbon Receptor Function: SNP-Dependent CYP1A1 Induction s Duan Liu, Sisi Qin, Balmiki Ray,1 Krishna R. Kalari, Liewei Wang, and Richard M. Weinshilboum Division of Clinical Pharmacology, Department of Molecular Pharmacology and Experimental Therapeutics (D.L., S.Q., B.R., L.W., R.M.W.) and Division of Biomedical Statistics and Informatics, Department of Health Sciences Research (K.R.K.), Mayo Clinic, Rochester, Minnesota Received April 22, 2018; accepted June 28, 2018 ABSTRACT Downloaded from CYP1A1 expression can be upregulated by the ligand-activated aryl fashion. LCLs with the AA genotype displayed significantly higher hydrocarbon receptor (AHR). Based on prior observations with AHR-XRE binding and CYP1A1 mRNA expression after 3MC estrogen receptors and estrogen response elements, we tested treatment than did those with the GG genotype. Electrophoretic the hypothesis that single-nucleotide polymorphisms (SNPs) map- mobility shift assay (EMSA) showed that oligonucleotides with the ping hundreds of base pairs (bp) from xenobiotic response elements AA genotype displayed higher LCL nuclear extract binding after (XREs) might influence AHR binding and subsequent gene expres- 3MC treatment than did those with the GG genotype, and mass dmd.aspetjournals.org sion. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Minichromosome Maintenance Complex Component 8 and 9 Gene Expression in the Menstrual Cycle and Unexplained Primary Ovarian Insufficiency
Journal of Assisted Reproduction and Genetics (2019) 36:57–64 https://doi.org/10.1007/s10815-018-1325-z GENETICS Minichromosome maintenance complex component 8 and 9 gene expression in the menstrual cycle and unexplained primary ovarian insufficiency Yelena Dondik1,2 & Zhenmin Lei1 & Jeremy Gaskins1 & Kelly Pagidas1 Received: 2 July 2018 /Accepted: 20 September 2018 /Published online: 1 October 2018 # Springer Science+Business Media, LLC, part of Springer Nature 2018 Abstract Purpose DNA repair genes Minichromosome maintenance complex component (MCM) 8 and 9 have been linked with gonadal development, primary ovarian insufficiency (POI), and age at menopause. Our objective was to characterize MCM 8 and 9 gene expression in the menstrual cycle, and to compare MCM 8/9 expression in POI vs normo-ovulatory women. Methods Normo-ovulatory controls (n = 11) and unexplained POI subjects (n = 6) were recruited. Controls provided three blood samples within one menstrual cycle: (1) early follicular phase, (2) ovulation, and (3) mid-luteal phase. Six of 11 controls only provided a follicular phase sample. Amenorrheic POI subjects provided a single, random blood sample. MCM8/9 expression in peripheral blood was assessed with qRTPCR. Analyses were performed using delta-Ct measurements; group differences were transformed to a fold change (FC) and confidence interval (CI). Differences across menstrual cycle phases were compared using random effects ANOVA. Two-sample t tests were used to compare two groups. Results MCM8 expression was significantly lower at ovulation and during the luteal phase, when compared to the follicular phase [FC = 0.69 in the luteal vs follicular phase (p = 0.012, CI = 0.53, 0.90); and 0.65 in the ovulatory vs follicular phase (p = 0.0057, CI = 0.50, 0.85)]. -

Genetics of Azoospermia
International Journal of Molecular Sciences Review Genetics of Azoospermia Francesca Cioppi , Viktoria Rosta and Csilla Krausz * Department of Biochemical, Experimental and Clinical Sciences “Mario Serio”, University of Florence, 50139 Florence, Italy; francesca.cioppi@unifi.it (F.C.); viktoria.rosta@unifi.it (V.R.) * Correspondence: csilla.krausz@unifi.it Abstract: Azoospermia affects 1% of men, and it can be due to: (i) hypothalamic-pituitary dysfunction, (ii) primary quantitative spermatogenic disturbances, (iii) urogenital duct obstruction. Known genetic factors contribute to all these categories, and genetic testing is part of the routine diagnostic workup of azoospermic men. The diagnostic yield of genetic tests in azoospermia is different in the different etiological categories, with the highest in Congenital Bilateral Absence of Vas Deferens (90%) and the lowest in Non-Obstructive Azoospermia (NOA) due to primary testicular failure (~30%). Whole- Exome Sequencing allowed the discovery of an increasing number of monogenic defects of NOA with a current list of 38 candidate genes. These genes are of potential clinical relevance for future gene panel-based screening. We classified these genes according to the associated-testicular histology underlying the NOA phenotype. The validation and the discovery of novel NOA genes will radically improve patient management. Interestingly, approximately 37% of candidate genes are shared in human male and female gonadal failure, implying that genetic counselling should be extended also to female family members of NOA patients. Keywords: azoospermia; infertility; genetics; exome; NGS; NOA; Klinefelter syndrome; Y chromosome microdeletions; CBAVD; congenital hypogonadotropic hypogonadism Citation: Cioppi, F.; Rosta, V.; Krausz, C. Genetics of Azoospermia. 1. Introduction Int. J. Mol. Sci. -

Comparative Transcriptomics Reveals Similarities and Differences
Seifert et al. BMC Cancer (2015) 15:952 DOI 10.1186/s12885-015-1939-9 RESEARCH ARTICLE Open Access Comparative transcriptomics reveals similarities and differences between astrocytoma grades Michael Seifert1,2,5*, Martin Garbe1, Betty Friedrich1,3, Michel Mittelbronn4 and Barbara Klink5,6,7 Abstract Background: Astrocytomas are the most common primary brain tumors distinguished into four histological grades. Molecular analyses of individual astrocytoma grades have revealed detailed insights into genetic, transcriptomic and epigenetic alterations. This provides an excellent basis to identify similarities and differences between astrocytoma grades. Methods: We utilized public omics data of all four astrocytoma grades focusing on pilocytic astrocytomas (PA I), diffuse astrocytomas (AS II), anaplastic astrocytomas (AS III) and glioblastomas (GBM IV) to identify similarities and differences using well-established bioinformatics and systems biology approaches. We further validated the expression and localization of Ang2 involved in angiogenesis using immunohistochemistry. Results: Our analyses show similarities and differences between astrocytoma grades at the level of individual genes, signaling pathways and regulatory networks. We identified many differentially expressed genes that were either exclusively observed in a specific astrocytoma grade or commonly affected in specific subsets of astrocytoma grades in comparison to normal brain. Further, the number of differentially expressed genes generally increased with the astrocytoma grade with one major exception. The cytokine receptor pathway showed nearly the same number of differentially expressed genes in PA I and GBM IV and was further characterized by a significant overlap of commonly altered genes and an exclusive enrichment of overexpressed cancer genes in GBM IV. Additional analyses revealed a strong exclusive overexpression of CX3CL1 (fractalkine) and its receptor CX3CR1 in PA I possibly contributing to the absence of invasive growth. -

Discovery of a Molecular Glue That Enhances Uprmt to Restore
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.17.431525; this version posted February 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Discovery of a molecular glue that enhances UPRmt to restore proteostasis via TRKA-GRB2-EVI1-CRLS1 axis Authors: Li-Feng-Rong Qi1, 2 †, Cheng Qian1, †, Shuai Liu1, 2†, Chao Peng3, 4, Mu Zhang1, Peng Yang1, Ping Wu3, 4, Ping Li1 and Xiaojun Xu1, 2 * † These authors share joint first authorship Running title: Ginsenoside Rg3 reverses Parkinson’s disease model by enhancing mitochondrial UPR Affiliations: 1 State Key Laboratory of Natural Medicines, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. 2 Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. 3. National Facility for Protein Science in Shanghai, Zhangjiang Lab, Shanghai Advanced Research Institute, Chinese Academy of Science, Shanghai 201210, China 4. Shanghai Science Research Center, Chinese Academy of Sciences, Shanghai, 201204, China. Corresponding author: Ping Li, State Key Laboratory of Natural Medicines, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. Email: [email protected], Xiaojun Xu, State Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. Telephone number: +86-2583271203, E-mail: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/2021.02.17.431525; this version posted February 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -
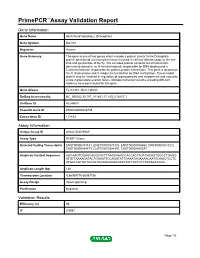
Download Validation Data
PrimePCR™Assay Validation Report Gene Information Gene Name dachshund homolog 2 (Drosophila) Gene Symbol DACH2 Organism Human Gene Summary This gene is one of two genes which encode a protein similar to the Drosophila protein dachshund a transcription factor involved in cell fate determination in the eye limb and genital disc of the fly. The encoded protein contains two characteristic dachshund domains: an N-terminal domain responsible for DNA binding and a C-terminal domain responsible for protein-protein interactions. This gene is located on the X chromosome and is subject to inactivation by DNA methylation. The encoded protein may be involved in regulation of organogenesis and myogenesis and may play a role in premature ovarian failure. Multiple transcript variants encoding different isoforms have been found for this gene. Gene Aliases FLJ31391, MGC138545 RefSeq Accession No. NC_000023.10, NT_011651.17, NG_012817.1 UniGene ID Hs.86603 Ensembl Gene ID ENSG00000126733 Entrez Gene ID 117154 Assay Information Unique Assay ID qHsaCID0009489 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000373131, ENST00000373125, ENST00000508860, ENST00000510272, ENST00000484479, ENST00000344497, ENST00000400297 Amplicon Context Sequence AGCAAGTGGAGCAGGCACTTAAGCAAGCCACCACTAGTGACAGTGGCCTGAGG ATGTTAAAAGATACTGGAATTCCAGATATTGAAATAGAAAACAATGGGACTCCTC ATGATAGTGCTGCTATGCAAGGAGGTAACTATTACTGTTTAGAAATGGC Amplicon Length (bp) 126 Chromosome Location X:86069775-86087136 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) -

Chromosomal Instability in Women with Primary Ovarian Insufficiency
Human Reproduction, Vol.33, No.3 pp. 531–538, 2018 Advanced Access publication on February 7, 2018 doi:10.1093/humrep/dey012 ORIGINAL ARTICLE Reproductive genetics Chromosomal instability in women with primary ovarian insufficiency † † Sunita Katari1,2, , Mahmoud Aarabi1,3, , Angela Kintigh3, Susan Mann3, Svetlana A. Yatsenko1,3,4,5,6, Joseph S. Sanfilippo1,2, Downloaded from https://academic.oup.com/humrep/article/33/3/531/4841816 by guest on 27 September 2021 Anthony J. Zeleznik2,6, and Aleksandar Rajkovic1,3,4,5,6,* 1Department of Obstetrics, Gynecology, and Reproductive Sciences, School of Medicine, University of Pittsburgh, 300 Halket Street, Pittsburgh, PA 15213, USA 2Division of Reproductive Endocrinology and Infertility, Magee-Womens Hospital of UPMC, 300 Halket Street, Pittsburgh, PA 15213, USA 3Medical Genetics & Genomics Laboratories, Magee Womens Hospital of UPMC, 300 Halket Street, Pittsburgh, PA 15213, USA 4Department of Pathology, School of Medicine, University of Pittsburgh, 200 Lothrop Street, Pittsburgh, PA 15261, USA 5Department of Human Genetics, School of Public Health, University of Pittsburgh, 130 De Soto Street, Pittsburgh, PA 15261, USA 6Magee Womens Research Institute, 204 Craft Avenue, Pittsburgh, PA 15213, USA *Correspondence address. Magee Womens Research Institute, 204 Craft Ave., A224, Pittsburgh, PA 15213, USA. E-mail: [email protected] Submitted on November 28, 2017; resubmitted on January 7, 2018; accepted on January 19, 2018 STUDY QUESTION: What is the prevalence of somatic chromosomal instability among women with idiopathic primary ovarian insuffi- ciency (POI)? SUMMARY ANSWER: A subset of women with idiopathic POI may have functional impairment in DNA repair leading to chromosomal instability in their soma. -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -

Loss of DACH2 Is a Candidate Early Event in Breast and Ovarian Carcinogenesis
Loss of DACH2 is a candidate early event in breast and ovarian carcinogenesis By Rania Chehade A thesis submitted in conformity with the requirements for Degree of Master of Science Medical Biophysics Department University of Toronto Copyright by Rania Chehade 2013 Loss of DACH2 is a candidate early event in breast and ovarian carcinogenesis Rania Chehade Master of Science Medical Biophysics Department University of Toronto 2013 Abstract Mechanistic insights into how enduring menstrual cycle hormonal signaling promotes tumorigenesis are emerging. We performed a genome-wide screen in primary epithelial cells to identify hormonally-regulated candidates that initiate pro-tumorigenic phenotypes in normal cells. One candidate, DACH2, has been described as part of a network that regulates organogenesis during development. In vitro, we find that DACH2 expression is regulated by estrogen and progesterone in a dosage dependent manner. Lentiviral-mediated shRNA silencing of DACH2 in hormonally responsive tissues promotes expansion of cells with progenitor characteristics. Within gene expression profiles of fallopian tubes, DACH2 is a member of a minimal gene classifier that distinguishes follicular versus luteal phases. Decreased DACH2 is characteristic of luteal-phase tubal cells and the majority of ovarian serous carcinomas. These studies suggest that DACH2 may orchestrate a physiological program that, when deregulated, locks cells in a progenitor-state, induces uncontrolled proliferation, and predisposes cells for breast and ovarian carcinogenesis. ii Acknowledgements Graduate school has been a bountiful journey of learning opportunities in academic, work and life relationships. I have developed various skills and I have matured both as a researcher and a human being. First and foremost, I would like to acknowledge my supervisor Dr. -

Hsmcm8 and Hsmcm9: Essential for Double-Strand Break Repair and Normal Ovarian Function
HsMCM8 and HsMCM9: Essential for Double-Strand Break Repair and Normal Ovarian Function by Elizabeth Paladin Jeffries Bachelor of Science, Indiana University of Pennsylvania, 2009 Submitted to the Graduate Faculty of The Kenneth P. Dietrich School of Arts & Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2015 UNIVERSITY OF PITTSBURGH The Kenneth P. Dietrich School of Arts & Sciences This dissertation was presented by Elizabeth P. Jeffries It was defended on May 4, 2015 and approved by Xinyu Liu, Assistant Professor, Department of Chemistry Aleksandar Rajkovic, Professor and Chair, Department of Obstetrics, Gynecology and Reproductive Sciences Dissertation Co-Advisor: Seth Horne, Associate Professor, Department of Chemisry Dissertation Co-Advisor: Michael Trakselis, Adjunct Associate Professor, Department of Chemistry, University of Pittsburgh ii HsMCM8 and HsMCM9: Essential for Double-Strand Break Repair and Normal Ovarian Function Elizabeth Paladin Jeffries, PhD University of Pittsburgh, 2015 Copyright © by Elizabeth P. Jeffries 2015 iii HsMCM8 AND HsMCM9: ESSENTIAL FOR DNA DOUBLE-STRAND BREAK REPAIR AND NORMAL OVARIAN FUNCTION Elizabeth Jeffries, PhD University of Pittsburgh, 2015 The minichromosome maintenance (MCM) family of proteins is conserved from archaea to humans, and its members have roles in initiating DNA replication. MCM8 and MCM9 are minimally characterized members of the eukaryotic MCM family that associate with one another and both contain conserved ATP binding and hydrolysis motifs. The MCM8-9 complex participates in repair of DNA double-strand breaks by homologous recombination, and MCM8 is implicated in meiotic recombination. We identified a novel alternatively spliced isoform of HsMCM9 that results in a medium length protein product (MCM9M) that eliminates a C-terminal extension of the fully spliced product (MCM9L).