A Novel Chromosomal Translocation Identified Due to Complex Genetic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Multistep Bioinformatic Approach Detects Putative Regulatory
BMC Bioinformatics BioMed Central Research article Open Access A multistep bioinformatic approach detects putative regulatory elements in gene promoters Stefania Bortoluzzi1, Alessandro Coppe1, Andrea Bisognin1, Cinzia Pizzi2 and Gian Antonio Danieli*1 Address: 1Department of Biology, University of Padova – Via Bassi 58/B, 35131, Padova, Italy and 2Department of Information Engineering, University of Padova – Via Gradenigo 6/B, 35131, Padova, Italy Email: Stefania Bortoluzzi - [email protected]; Alessandro Coppe - [email protected]; Andrea Bisognin - [email protected]; Cinzia Pizzi - [email protected]; Gian Antonio Danieli* - [email protected] * Corresponding author Published: 18 May 2005 Received: 12 November 2004 Accepted: 18 May 2005 BMC Bioinformatics 2005, 6:121 doi:10.1186/1471-2105-6-121 This article is available from: http://www.biomedcentral.com/1471-2105/6/121 © 2005 Bortoluzzi et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Searching for approximate patterns in large promoter sequences frequently produces an exceedingly high numbers of results. Our aim was to exploit biological knowledge for definition of a sheltered search space and of appropriate search parameters, in order to develop a method for identification of a tractable number of sequence motifs. Results: Novel software (COOP) was developed for extraction of sequence motifs, based on clustering of exact or approximate patterns according to the frequency of their overlapping occurrences. -

Snps) Distant from Xenobiotic Response Elements Can Modulate Aryl Hydrocarbon Receptor Function: SNP-Dependent CYP1A1 Induction S
Supplemental material to this article can be found at: http://dmd.aspetjournals.org/content/suppl/2018/07/06/dmd.118.082164.DC1 1521-009X/46/9/1372–1381$35.00 https://doi.org/10.1124/dmd.118.082164 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 46:1372–1381, September 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Single Nucleotide Polymorphisms (SNPs) Distant from Xenobiotic Response Elements Can Modulate Aryl Hydrocarbon Receptor Function: SNP-Dependent CYP1A1 Induction s Duan Liu, Sisi Qin, Balmiki Ray,1 Krishna R. Kalari, Liewei Wang, and Richard M. Weinshilboum Division of Clinical Pharmacology, Department of Molecular Pharmacology and Experimental Therapeutics (D.L., S.Q., B.R., L.W., R.M.W.) and Division of Biomedical Statistics and Informatics, Department of Health Sciences Research (K.R.K.), Mayo Clinic, Rochester, Minnesota Received April 22, 2018; accepted June 28, 2018 ABSTRACT Downloaded from CYP1A1 expression can be upregulated by the ligand-activated aryl fashion. LCLs with the AA genotype displayed significantly higher hydrocarbon receptor (AHR). Based on prior observations with AHR-XRE binding and CYP1A1 mRNA expression after 3MC estrogen receptors and estrogen response elements, we tested treatment than did those with the GG genotype. Electrophoretic the hypothesis that single-nucleotide polymorphisms (SNPs) map- mobility shift assay (EMSA) showed that oligonucleotides with the ping hundreds of base pairs (bp) from xenobiotic response elements AA genotype displayed higher LCL nuclear extract binding after (XREs) might influence AHR binding and subsequent gene expres- 3MC treatment than did those with the GG genotype, and mass dmd.aspetjournals.org sion. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Comparative Transcriptomics Reveals Similarities and Differences
Seifert et al. BMC Cancer (2015) 15:952 DOI 10.1186/s12885-015-1939-9 RESEARCH ARTICLE Open Access Comparative transcriptomics reveals similarities and differences between astrocytoma grades Michael Seifert1,2,5*, Martin Garbe1, Betty Friedrich1,3, Michel Mittelbronn4 and Barbara Klink5,6,7 Abstract Background: Astrocytomas are the most common primary brain tumors distinguished into four histological grades. Molecular analyses of individual astrocytoma grades have revealed detailed insights into genetic, transcriptomic and epigenetic alterations. This provides an excellent basis to identify similarities and differences between astrocytoma grades. Methods: We utilized public omics data of all four astrocytoma grades focusing on pilocytic astrocytomas (PA I), diffuse astrocytomas (AS II), anaplastic astrocytomas (AS III) and glioblastomas (GBM IV) to identify similarities and differences using well-established bioinformatics and systems biology approaches. We further validated the expression and localization of Ang2 involved in angiogenesis using immunohistochemistry. Results: Our analyses show similarities and differences between astrocytoma grades at the level of individual genes, signaling pathways and regulatory networks. We identified many differentially expressed genes that were either exclusively observed in a specific astrocytoma grade or commonly affected in specific subsets of astrocytoma grades in comparison to normal brain. Further, the number of differentially expressed genes generally increased with the astrocytoma grade with one major exception. The cytokine receptor pathway showed nearly the same number of differentially expressed genes in PA I and GBM IV and was further characterized by a significant overlap of commonly altered genes and an exclusive enrichment of overexpressed cancer genes in GBM IV. Additional analyses revealed a strong exclusive overexpression of CX3CL1 (fractalkine) and its receptor CX3CR1 in PA I possibly contributing to the absence of invasive growth. -

University of London Thesis
REFERENCE ONLY UNIVERSITY OF LONDON THESIS Degree pWvO Y e a r Name of Author ^ COPYRIGHT This is a thesis accepted for a Higher Degree of the University of London. It is an unpublished typescript and the copyright is held by the author. All persons consulting the thesis must read and abide by the Copyright Declaration below. COPYRIGHT DECLARATION I recognise that the copyright of the above-described thesis rests with the author and that no quotation from it or information derived from it may be published without the prior written consent of the author. LOANS Theses may not be lent to individuals, but the Senate House Library may lend a copy to approved libraries within the United Kingdom, for consultation solely on the premises of those libraries. Application should be made to: Inter-Library Loans, Senate House Library, Senate House, Malet Street, London WC1E 7HU. REPRODUCTION University of London theses may not be reproduced without explicit written permission from the Senate House Library. Enquiries should be addressed to the Theses Section of the Library. Regulations concerning reproduction vary according to the date of acceptance of the thesis and are listed below as guidelines. A. Before 1962. Permission granted only upon the prior written consent of the author. (The Senate House Library will provide addresses where possible). B. 1962 - 1974. In many cases the author has agreed to permit copying upon completion of a Copyright Declaration. C. 1975 - 1988. Most theses may be copied upon completion of a Copyright Declaration. D. 1989 onwards. Most theses may be copied. This thesis comes within category D. -

Discovery of a Molecular Glue That Enhances Uprmt to Restore
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.17.431525; this version posted February 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Discovery of a molecular glue that enhances UPRmt to restore proteostasis via TRKA-GRB2-EVI1-CRLS1 axis Authors: Li-Feng-Rong Qi1, 2 †, Cheng Qian1, †, Shuai Liu1, 2†, Chao Peng3, 4, Mu Zhang1, Peng Yang1, Ping Wu3, 4, Ping Li1 and Xiaojun Xu1, 2 * † These authors share joint first authorship Running title: Ginsenoside Rg3 reverses Parkinson’s disease model by enhancing mitochondrial UPR Affiliations: 1 State Key Laboratory of Natural Medicines, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. 2 Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. 3. National Facility for Protein Science in Shanghai, Zhangjiang Lab, Shanghai Advanced Research Institute, Chinese Academy of Science, Shanghai 201210, China 4. Shanghai Science Research Center, Chinese Academy of Sciences, Shanghai, 201204, China. Corresponding author: Ping Li, State Key Laboratory of Natural Medicines, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. Email: [email protected], Xiaojun Xu, State Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China. Telephone number: +86-2583271203, E-mail: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/2021.02.17.431525; this version posted February 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Towards the Identification of Causal Genes for Age-Related Macular
bioRxiv preprint doi: https://doi.org/10.1101/778613; this version posted September 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Towards the identification of causal genes for age-related macular degeneration 2 3 Fei-Fei Cheng1,2,3,5, You-Yuan Zhuang1,2,5, Xin-Ran Wen1,2, Angli Xue4, Jian Yang3,4,6,*, Zi-Bing 4 Jin1,2,6,* 5 6 1Division of Ophthalmic Genetics, The Eye Hospital, Wenzhou Medical University, Wenzhou 325027, 7 China; 8 2National Center for International Research in Regenerative Medicine and Neurogenetics, National 9 Clinical Research Center for Ophthalmology, State Key Laboratory of Ophthalmology, Optometry and 10 Visual Science, Wenzhou, 325027 China; 11 3Institute for Advanced Research, Wenzhou Medical University, Wenzhou 325035, China; 12 4Institute for Molecular Bioscience, The University of Queensland, Brisbane, Queensland, 4072, 13 Australia; 14 5These authors contributed equally. 15 6These authors jointly supervised this work. 16 *Correspondence: Zi-Bing Jin <[email protected]> or Jian Yang <[email protected]>. 17 18 Abstract 19 Age-related macular degeneration (AMD) is a leading cause of visual impairment in ageing 20 populations and has no radical treatment or prevention. Although genome-wide association studies 21 (GWAS) have identified many susceptibility loci for AMD, the underlying causal genes remain 22 elusive. Here, we prioritized nine putative causal genes by integrating expression quantitative trait 23 locus (eQTL) data from blood (푛 = 2,765) with AMD GWAS data (16,144 cases vs. -
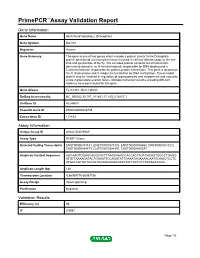
Download Validation Data
PrimePCR™Assay Validation Report Gene Information Gene Name dachshund homolog 2 (Drosophila) Gene Symbol DACH2 Organism Human Gene Summary This gene is one of two genes which encode a protein similar to the Drosophila protein dachshund a transcription factor involved in cell fate determination in the eye limb and genital disc of the fly. The encoded protein contains two characteristic dachshund domains: an N-terminal domain responsible for DNA binding and a C-terminal domain responsible for protein-protein interactions. This gene is located on the X chromosome and is subject to inactivation by DNA methylation. The encoded protein may be involved in regulation of organogenesis and myogenesis and may play a role in premature ovarian failure. Multiple transcript variants encoding different isoforms have been found for this gene. Gene Aliases FLJ31391, MGC138545 RefSeq Accession No. NC_000023.10, NT_011651.17, NG_012817.1 UniGene ID Hs.86603 Ensembl Gene ID ENSG00000126733 Entrez Gene ID 117154 Assay Information Unique Assay ID qHsaCID0009489 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000373131, ENST00000373125, ENST00000508860, ENST00000510272, ENST00000484479, ENST00000344497, ENST00000400297 Amplicon Context Sequence AGCAAGTGGAGCAGGCACTTAAGCAAGCCACCACTAGTGACAGTGGCCTGAGG ATGTTAAAAGATACTGGAATTCCAGATATTGAAATAGAAAACAATGGGACTCCTC ATGATAGTGCTGCTATGCAAGGAGGTAACTATTACTGTTTAGAAATGGC Amplicon Length (bp) 126 Chromosome Location X:86069775-86087136 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) -

Genetic Analyses of Human Fetal Retinal Pigment Epithelium Gene Expression Suggest Ocular Disease Mechanisms
ARTICLE https://doi.org/10.1038/s42003-019-0430-6 OPEN Genetic analyses of human fetal retinal pigment epithelium gene expression suggest ocular disease mechanisms Boxiang Liu 1,6, Melissa A. Calton2,6, Nathan S. Abell2, Gillie Benchorin2, Michael J. Gloudemans 3, 1234567890():,; Ming Chen2, Jane Hu4, Xin Li 5, Brunilda Balliu5, Dean Bok4, Stephen B. Montgomery 2,5 & Douglas Vollrath2 The retinal pigment epithelium (RPE) serves vital roles in ocular development and retinal homeostasis but has limited representation in large-scale functional genomics datasets. Understanding how common human genetic variants affect RPE gene expression could elu- cidate the sources of phenotypic variability in selected monogenic ocular diseases and pin- point causal genes at genome-wide association study (GWAS) loci. We interrogated the genetics of gene expression of cultured human fetal RPE (fRPE) cells under two metabolic conditions and discovered hundreds of shared or condition-specific expression or splice quantitative trait loci (e/sQTLs). Co-localizations of fRPE e/sQTLs with age-related macular degeneration (AMD) and myopia GWAS data suggest new candidate genes, and mechan- isms by which a common RDH5 allele contributes to both increased AMD risk and decreased myopia risk. Our study highlights the unique transcriptomic characteristics of fRPE and provides a resource to connect e/sQTLs in a critical ocular cell type to monogenic and complex eye disorders. 1 Department of Biology, Stanford University, Stanford, CA 94305, USA. 2 Department of Genetics, Stanford University School of Medicine, Stanford, CA 94305, USA. 3 Program in Biomedical Informatics, Stanford University School of Medicine, Stanford 94305 CA, USA. 4 Department of Ophthalmology, Jules Stein Eye Institute, UCLA, Los Angeles 90095 CA, USA. -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -

A Large Genome-Wide Association Study of Age-Related Macular Degeneration Highlights Contributions of Rare and Common Variants
A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Lars G. Fritsche1†, Wilmar Igl2†, Jessica N. Cooke Bailey3†, Felix Grassmann4†, Sebanti Sengupta1†, Jennifer L. Bragg-Gresham1,5, Kathryn P. Burdon6, Scott J. Hebbring7, Cindy Wen8, Mathias Gorski2, Ivana K. Kim9, David Cho10, Donald Zack11,12,13,14,15, Eric Souied16, Hendrik P. N. Scholl11,17, Elisa Bala18, Kristine E. Lee19, David J. Hunter20,21, Rebecca J. Sardell22, Paul Mitchell23, Joanna E. Merriam24, Valentina Cipriani25,26, Joshua D. Hoffman27, Tina Schick28, Yara T. E. Lechanteur29, Robyn H. Guymer30, Matthew P. Johnson31, Yingda Jiang32, Chloe M. Stanton33, Gabriëlle H. S. Buitendijk34,35, Xiaowei Zhan1,36,37, Alan M. Kwong1, Alexis Boleda38, Matthew Brooks39, Linn Gieser38, Rinki Ratnapriya38, Kari E. Branham39, Johanna R. Foerster1, John R. Heckenlively39, Mohammad I. Othman39, Brendan J. Vote6, Helena Hai Liang30, Emmanuelle Souzeau40, Ian L. McAllister41, Timothy Isaacs41, Janette Hall40, Stewart Lake40, David A. Mackey6,30,41, Ian J. Constable41, Jamie E. Craig40, Terrie E. Kitchner7, Zhenglin Yang42,43, Zhiguang Su44, Hongrong Luo8,44, Daniel Chen8, Hong Ouyang8, Ken Flagg8, Danni Lin8, Guanping Mao8, Henry Ferreyra8, Klaus Stark2, Claudia N. von Strachwitz45, Armin Wolf46, Caroline Brandl2,4,47, Guenther Rudolph46, Matthias Olden2, Margaux A. Morrison48, Denise J. Morgan48, Matthew Schu49,50,51,52,53, Jeeyun Ahn54, Giuliana Silvestri55, Evangelia E. Tsironi56, Kyu Hyung Park57, Lindsay A. Farrer49,50,51,52,53, Anton Orlin58, Alexander Brucker59, Mingyao Li60, Christine Curcio61, Saddek Mohand-Saïd62,63,64,65, José-Alain Sahel62,63,64,65,66,67,68, Isabelle Audo62,63,64,69, Mustapha Benchaboune65, Angela J.