(TPOXX®), the First Antiviral Against Smallpox
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Infectious Diseases
2013 MEDICINES IN DEVELOPMENT REPORT Infectious Diseases A Report on Diseases Caused by Bacteria, Viruses, Fungi and Parasites PRESENTED BY AMERICA’S BIOPHARMACEUTICAL RESEARCH COMPANIES Biopharmaceutical Research Evolves Against Infectious Diseases with Nearly 400 Medicines and Vaccines in Testing Throughout history, infectious diseases hepatitis C that inhibits the enzyme have taken a devastating toll on the lives essential for viral replication. and well-being of people around the • An anti-malarial drug that has shown Medicines in Development world. Caused when pathogens such activity against Plasmodium falci- For Infectious Diseases as bacteria or viruses enter a body and parum malaria which is resistant to multiply, infectious diseases were the current treatments. Application leading cause of death in the United Submitted States until the 1920s. Today, vaccines • A potential new antibiotic to treat methicillin-resistant Staphylococcus Phase III and infectious disease treatments have proven to be effective treatments in aureus (MRSA). Phase II many cases, but infectious diseases still • A novel treatment that works by Phase I pose a very serious threat to patients. blocking the ability of the smallpox Recently, some infectious pathogens, virus to spread to other cells, thus 226 such as pseudomonas bacteria, have preventing it from causing disease. become resistant to available treatments. Infectious diseases may never be fully Diseases once considered conquered, eradicated. However, new knowledge, such as tuberculosis, have reemerged new technologies, and the continuing as a growing health threat. commitment of America’s biopharma- America’s biopharmaceutical research ceutical research companies can help companies are developing 394 medicines meet the continuing—and ever-changing and vaccines to combat the many threats —threat from infectious diseases. -
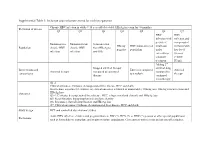
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

AHFS Pharmacologic-Therapeutic Classification System
AHFS Pharmacologic-Therapeutic Classification System Abacavir 48:24 - Mucolytic Agents - 382638 8:18.08.20 - HIV Nucleoside and Nucleotide Reverse Acitretin 84:92 - Skin and Mucous Membrane Agents, Abaloparatide 68:24.08 - Parathyroid Agents - 317036 Aclidinium Abatacept 12:08.08 - Antimuscarinics/Antispasmodics - 313022 92:36 - Disease-modifying Antirheumatic Drugs - Acrivastine 92:20 - Immunomodulatory Agents - 306003 4:08 - Second Generation Antihistamines - 394040 Abciximab 48:04.08 - Second Generation Antihistamines - 394040 20:12.18 - Platelet-aggregation Inhibitors - 395014 Acyclovir Abemaciclib 8:18.32 - Nucleosides and Nucleotides - 381045 10:00 - Antineoplastic Agents - 317058 84:04.06 - Antivirals - 381036 Abiraterone Adalimumab; -adaz 10:00 - Antineoplastic Agents - 311027 92:36 - Disease-modifying Antirheumatic Drugs - AbobotulinumtoxinA 56:92 - GI Drugs, Miscellaneous - 302046 92:20 - Immunomodulatory Agents - 302046 92:92 - Other Miscellaneous Therapeutic Agents - 12:20.92 - Skeletal Muscle Relaxants, Miscellaneous - Adapalene 84:92 - Skin and Mucous Membrane Agents, Acalabrutinib 10:00 - Antineoplastic Agents - 317059 Adefovir Acamprosate 8:18.32 - Nucleosides and Nucleotides - 302036 28:92 - Central Nervous System Agents, Adenosine 24:04.04.24 - Class IV Antiarrhythmics - 304010 Acarbose Adenovirus Vaccine Live Oral 68:20.02 - alpha-Glucosidase Inhibitors - 396015 80:12 - Vaccines - 315016 Acebutolol Ado-Trastuzumab 24:24 - beta-Adrenergic Blocking Agents - 387003 10:00 - Antineoplastic Agents - 313041 12:16.08.08 - Selective -

Keeping up with FDA Drug Approvals: 60 New Drugs in 60 Minutes Elizabeth A
Keeping Up with FDA Drug Approvals: 60 New Drugs in 60 Minutes Elizabeth A. Shlom, PharmD, BCPS Senior Vice President & Director Clinical Pharmacy Program | Acurity, Inc. Privileged and Confidential April 10, 2019 Privileged and Confidential Program Objectives By the end of the presentation, the pharmacist or pharmacy technician participant will be able to: ▪ Identify orphan drugs and first-in-class medications approved by the FDA in 2018. ▪ List five new drugs and their indications. ▪ Identify the place in therapy for three novel monoclonal antibodies. ▪ Discuss at least two new medications that address public health concerns. Dr. Shlom does not have any conflicts of interest in regard to this presentation. Both trade names and generic names will be discussed throughout the presentation Privileged and Confidential 2018 NDA Approvals (NMEs/BLAs) ▪ Lutathera (lutetium Lu 177 dotatate) ▪ Braftovi (encorafenib) ▪ Vizimpro (dacomitinib) ▪ Biktarvy (bictegravir, emtricitabine, ▪ TPOXX (tecovirimat) ▪ Libtayo (cemiplimab-rwic) tenofovir, ▪ Tibsovo (ivosidenib) ▪ Seysara (sarecycline) alafenamide) ▪ Krintafel (tafenoquine) ▪ Nuzyra (omadacycline) ▪ Symdeko (tezacaftor, ivacaftor) ▪ Orilissa (elagolix sodium) ▪ Revcovi (elapegademase-lvir) ▪ Erleada (apalutamide) ▪ Omegaven (fish oil triglycerides) ▪ Tegsedi (inotersen) ▪ Trogarzo (ibalizumab-uiyk) ▪ Mulpleta (lusutrombopag) ▪ Talzenna (talazoparib) ▪ Ilumya (tildrakizumab-asmn) ▪ Poteligeo (mogamulizumab-kpkc) ▪ Xofluza (baloxavir marboxil) ▪ Tavalisse (fostamatinib disodium) ▪ Onpattro (patisiran) -

The Role of Brincidofovir in Preparation for a Potential Smallpox Outbreak
viruses Perspective The Role of Brincidofovir in Preparation for a Potential Smallpox Outbreak Scott A. Foster 1,*, Scott Parker 2 ID and Randall Lanier 1 1 Chimerix, Durham, NC 27713, USA; [email protected] 2 Department of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, St. Louis, MO 63104, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-919-597-7741 Received: 29 September 2017; Accepted: 26 October 2017; Published: 30 October 2017 Abstract: Smallpox (variola) virus is considered a Category A bioterrorism agent due to its ability to spread rapidly and the high morbidity and mortality rates associated with infection. Current recommendations recognize the importance of oral antivirals and call for having at least two smallpox antivirals with different mechanisms of action available in the event of a smallpox outbreak. Multiple antivirals are recommended due in large part to the propensity of viruses to become resistant to antiviral therapy, especially monotherapy. Advances in synthetic biology heighten concerns that a bioterror attack with variola would utilize engineered resistance to antivirals and potentially vaccines. Brincidofovir, an oral antiviral in late stage development, has proven effective against orthopoxviruses in vitro and in vivo, has a different mechanism of action from tecovirimat (the only oral smallpox antiviral currently in the US Strategic National Stockpile), and has a resistance profile that reduces concerns in the scenario of a bioterror attack using genetically engineered smallpox. Given the devastating potential of smallpox as a bioweapon, preparation of a multi-pronged defense that accounts for the most obvious bioengineering possibilities is strategically imperative. Keywords: smallpox; variola virus; bioterrorism; bioweapon; brincidofovir; CMX001; antiviral 1. -

Comparison of Host Cell Gene Expression in Cowpox, Monkeypox Or Vaccinia Virus-Infected Cells Reveals Virus-Specific Regulation
Bourquain et al. Virology Journal 2013, 10:61 http://www.virologyj.com/content/10/1/61 RESEARCH Open Access Comparison of host cell gene expression in cowpox, monkeypox or vaccinia virus-infected cells reveals virus-specific regulation of immune response genes Daniel Bourquain1†, Piotr Wojtek Dabrowski1,2† and Andreas Nitsche1* Abstract Background: Animal-borne orthopoxviruses, like monkeypox, vaccinia and the closely related cowpox virus, are all capable of causing zoonotic infections in humans, representing a potential threat to human health. The disease caused by each virus differs in terms of symptoms and severity, but little is yet know about the reasons for these varying phenotypes. They may be explained by the unique repertoire of immune and host cell modulating factors encoded by each virus. In this study, we analysed the specific modulation of the host cell’s gene expression profile by cowpox, monkeypox and vaccinia virus infection. We aimed to identify mechanisms that are either common to orthopoxvirus infection or specific to certain orthopoxvirus species, allowing a more detailed description of differences in virus-host cell interactions between individual orthopoxviruses. To this end, we analysed changes in host cell gene expression of HeLa cells in response to infection with cowpox, monkeypox and vaccinia virus, using whole-genome gene expression microarrays, and compared these to each other and to non-infected cells. Results: Despite a dominating non-responsiveness of cellular transcription towards orthopoxvirus infection, we could identify several clusters of infection-modulated genes. These clusters are either commonly regulated by orthopoxvirus infection or are uniquely regulated by infection with a specific orthopoxvirus, with major differences being observed in immune response genes. -

2019 Icar Program & Abstracts Book
Hosted by the International Society for Antiviral Research (ISAR) ND International Conference 32on Antiviral Research (ICAR) Baltimore MARYLAND PROGRAM and USA Hyatt Regency BALTIMORE ABSTRACTS May 12-15 2019 ND TABLE OF International Conference CONTENTS 32on Antiviral Research (ICAR) Daily Schedule . .3 Organization . 4 Contributors . 5 Keynotes & Networking . 6 Schedule at a Glance . 7 ISAR Awardees . 10 The 2019 Chu Family Foundation Scholarship Awardees . 15 Speaker Biographies . 17 Program Schedule . .25 Posters . 37 Abstracts . 53 Author Index . 130 PROGRAM and ABSTRACTS of the 32nd International Conference on Antiviral Research (ICAR) 2 ND DAILY International Conference SCHEDULE 32on Antiviral Research (ICAR) SUNDAY, MAY 12, 2019 › Women in Science Roundtable › Welcome and Keynote Lectures › Antonín Holý Memorial Award Lecture › Influenza Symposium › Opening Reception MONDAY, MAY 13, 2019 › Women in Science Award Lecture › Emerging Virus Symposium › Short Presentations 1 › Poster Session 1 › Retrovirus Symposium › ISAR Award of Excellence Presentation › PechaKucha Event with Introduction of First Time Attendees TUESDAY, MAY 14, 2019 › What’s New in Antiviral Research 1 › Short Presentations 2 & 3 › ISAR Award for Outstanding Contributions to the Society Presentation › Career Development Panel › William Prusoff Young Investigator Award Lecture › Medicinal Chemistry Symposium › Poster Session 2 › Networking Reception WEDNESDAY, MAY 15, 2019 › Gertrude Elion Memorial Award Lecture › What’s New in Antiviral Research 2 › Shotgun Oral -

Synthesis of Polycyclic Compounds with Antiviral Activity
Synthesis of polycyclic compounds with antiviral activity Eva Torres Costa ADVERTIMENT. La consulta d’aquesta tesi queda condicionada a l’acceptació de les següents condicions d'ús: La difusió d’aquesta tesi per mitjà del servei TDX (www.tdx.cat) i a través del Dipòsit Digital de la UB (diposit.ub.edu) ha estat autoritzada pels titulars dels drets de propietat intel·lectual únicament per a usos privats emmarcats en activitats d’investigació i docència. No s’autoritza la seva reproducció amb finalitats de lucre ni la seva difusió i posada a disposició des d’un lloc aliè al servei TDX ni al Dipòsit Digital de la UB. No s’autoritza la presentació del seu contingut en una finestra o marc aliè a TDX o al Dipòsit Digital de la UB (framing). Aquesta reserva de drets afecta tant al resum de presentació de la tesi com als seus continguts. En la utilització o cita de parts de la tesi és obligat indicar el nom de la persona autora. ADVERTENCIA. La consulta de esta tesis queda condicionada a la aceptación de las siguientes condiciones de uso: La difusión de esta tesis por medio del servicio TDR (www.tdx.cat) y a través del Repositorio Digital de la UB (diposit.ub.edu) ha sido autorizada por los titulares de los derechos de propiedad intelectual únicamente para usos privados enmarcados en actividades de investigación y docencia. No se autoriza su reproducción con finalidades de lucro ni su difusión y puesta a disposición desde un sitio ajeno al servicio TDR o al Repositorio Digital de la UB. -

Drug Pipeline Monthly Update
Drug Pipeline Monthly Update Critical updates in an ever changing environment December 2018 New drug information ● Temixys™ (lamivudine/tenofovir disoproxil fumarate): The Food and Drug Administration (FDA) approved Celltrion’s Temixys in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 35 kg, dosed once daily. Temixys is a combination of two nucleoside reverse transcriptase inhibitors (NRTIs) in one tablet. Celltrion intends to sell Temixys at a discounted price compared to Gilead’s Truvada® (emtricitabine/tenofovir disoproxil fumarate).1 Celltrion plans to launch early 2019.1 ● Tolsura™ (itraconazole) capsules: Mayne Pharma received FDA approval for Tolsura for the treatment of systemic fungal infections in adult patients including certain forms of: → blastomycosis, → histoplasmosis, and → aspergillosis Mayne notes that this is a Super-BioAvailable (SUBA) formulation of itraconazole that has been shown in clinical studies to have increased bioavailability and reduced variability when compared to other itraconazole formulations.2 Tolsura will launch in January 2019.2 ● Montegrity™ (prucalopride): The FDA approved Shire’s Montegrity, a once daily, oral medication for the treatment of adults with chronic idiopathic constipation (CIC). Montegrity will compete for market share in the CIC space with Allergan’s Linzess® (linaclotide), Takeda’s Amitiza® (lubiprostone), and Synergy’s Trulance® (plecanatide). Montegrity is anticipated to launch in 2019. continued While the information in this newsletter is from sources we believe to be reliable, we do not warrant that the information in this document is free from error. Use it only as a guide. Statements regarding drugs or manufacturers are not intended as promotion; those statements should not be used to make assumptions about formulary status. -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

Attenuation of B5R Mutants of Rabbitpox Virus in Vivo Is Related
VIROLOGY 233, 118–129 (1997) ARTICLE NO. VY978556 View metadata, citation and similar papers at core.ac.uk brought to you by CORE Attenuation of B5R Mutants of Rabbitpox Virus in Vivo Is Related to Impaired Growth provided by Elsevier - Publisher Connector and Not an Enhanced Host Inflammatory Response Richard J. Stern,* James P. Thompson,† and R. W. Moyer*,1 *Department of Molecular Genetics and Microbiology University of Florida College of Medicine, Gainesville, Florida 32610-0266; and †Department of Small Animal Clinical Sciences, College of Veterinary Medicine, Gainesville, Florida 32610-0216 Received December 23, 1996; accepted March 21, 1997 The rabbitpox virus (RPV) B5R protein, synthesized late in infection, is found as a 45-kDa membrane-associated protein of the envelope of infectious extracellular enveloped virus (EEV) and as a 38-kDa protein secreted from the cell by a process independent of morphogenesis. The protein is not found associated with intracellular mature virus (IMV). Deletion of the gene attenuates the virus (RPVDB5R) in animals (mice and rabbits), has relatively little effect on formation of IMV, prevents EEV formation in some but not all cells, and leads to a reduced host range. Analysis of the sequence of the protein suggests relatedness to factor H of the complement cascade. Collectively, these observations suggest that attenuation of the virus in vivo could be linked to an inhibition of the inflammatory response, a deficiency in growth, or both. In this report we have analyzed the behavior of RPVDB5R in infected mice and rabbits and conclude that attenuation of the mutant virus likely results from simple failure to grow within the infected animal and that the inflammatory response probably contributes little to the observed attenuation. -

Here, There, and Everywhere: the Wide Host Range and Geographic Distribution of Zoonotic Orthopoxviruses
viruses Review Here, There, and Everywhere: The Wide Host Range and Geographic Distribution of Zoonotic Orthopoxviruses Natalia Ingrid Oliveira Silva, Jaqueline Silva de Oliveira, Erna Geessien Kroon , Giliane de Souza Trindade and Betânia Paiva Drumond * Laboratório de Vírus, Departamento de Microbiologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais: Belo Horizonte, Minas Gerais 31270-901, Brazil; [email protected] (N.I.O.S.); [email protected] (J.S.d.O.); [email protected] (E.G.K.); [email protected] (G.d.S.T.) * Correspondence: [email protected] Abstract: The global emergence of zoonotic viruses, including poxviruses, poses one of the greatest threats to human and animal health. Forty years after the eradication of smallpox, emerging zoonotic orthopoxviruses, such as monkeypox, cowpox, and vaccinia viruses continue to infect humans as well as wild and domestic animals. Currently, the geographical distribution of poxviruses in a broad range of hosts worldwide raises concerns regarding the possibility of outbreaks or viral dissemination to new geographical regions. Here, we review the global host ranges and current epidemiological understanding of zoonotic orthopoxviruses while focusing on orthopoxviruses with epidemic potential, including monkeypox, cowpox, and vaccinia viruses. Keywords: Orthopoxvirus; Poxviridae; zoonosis; Monkeypox virus; Cowpox virus; Vaccinia virus; host range; wild and domestic animals; emergent viruses; outbreak Citation: Silva, N.I.O.; de Oliveira, J.S.; Kroon, E.G.; Trindade, G.d.S.; Drumond, B.P. Here, There, and Everywhere: The Wide Host Range 1. Poxvirus and Emerging Diseases and Geographic Distribution of Zoonotic diseases, defined as diseases or infections that are naturally transmissible Zoonotic Orthopoxviruses. Viruses from vertebrate animals to humans, represent a significant threat to global health [1].