Bhiva Guidelines on the Use of Vaccines in Hiv-Positive Adults 2015
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Infectious Diseases
2013 MEDICINES IN DEVELOPMENT REPORT Infectious Diseases A Report on Diseases Caused by Bacteria, Viruses, Fungi and Parasites PRESENTED BY AMERICA’S BIOPHARMACEUTICAL RESEARCH COMPANIES Biopharmaceutical Research Evolves Against Infectious Diseases with Nearly 400 Medicines and Vaccines in Testing Throughout history, infectious diseases hepatitis C that inhibits the enzyme have taken a devastating toll on the lives essential for viral replication. and well-being of people around the • An anti-malarial drug that has shown Medicines in Development world. Caused when pathogens such activity against Plasmodium falci- For Infectious Diseases as bacteria or viruses enter a body and parum malaria which is resistant to multiply, infectious diseases were the current treatments. Application leading cause of death in the United Submitted States until the 1920s. Today, vaccines • A potential new antibiotic to treat methicillin-resistant Staphylococcus Phase III and infectious disease treatments have proven to be effective treatments in aureus (MRSA). Phase II many cases, but infectious diseases still • A novel treatment that works by Phase I pose a very serious threat to patients. blocking the ability of the smallpox Recently, some infectious pathogens, virus to spread to other cells, thus 226 such as pseudomonas bacteria, have preventing it from causing disease. become resistant to available treatments. Infectious diseases may never be fully Diseases once considered conquered, eradicated. However, new knowledge, such as tuberculosis, have reemerged new technologies, and the continuing as a growing health threat. commitment of America’s biopharma- America’s biopharmaceutical research ceutical research companies can help companies are developing 394 medicines meet the continuing—and ever-changing and vaccines to combat the many threats —threat from infectious diseases. -
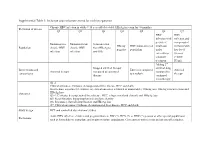
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

AHFS Pharmacologic-Therapeutic Classification System
AHFS Pharmacologic-Therapeutic Classification System Abacavir 48:24 - Mucolytic Agents - 382638 8:18.08.20 - HIV Nucleoside and Nucleotide Reverse Acitretin 84:92 - Skin and Mucous Membrane Agents, Abaloparatide 68:24.08 - Parathyroid Agents - 317036 Aclidinium Abatacept 12:08.08 - Antimuscarinics/Antispasmodics - 313022 92:36 - Disease-modifying Antirheumatic Drugs - Acrivastine 92:20 - Immunomodulatory Agents - 306003 4:08 - Second Generation Antihistamines - 394040 Abciximab 48:04.08 - Second Generation Antihistamines - 394040 20:12.18 - Platelet-aggregation Inhibitors - 395014 Acyclovir Abemaciclib 8:18.32 - Nucleosides and Nucleotides - 381045 10:00 - Antineoplastic Agents - 317058 84:04.06 - Antivirals - 381036 Abiraterone Adalimumab; -adaz 10:00 - Antineoplastic Agents - 311027 92:36 - Disease-modifying Antirheumatic Drugs - AbobotulinumtoxinA 56:92 - GI Drugs, Miscellaneous - 302046 92:20 - Immunomodulatory Agents - 302046 92:92 - Other Miscellaneous Therapeutic Agents - 12:20.92 - Skeletal Muscle Relaxants, Miscellaneous - Adapalene 84:92 - Skin and Mucous Membrane Agents, Acalabrutinib 10:00 - Antineoplastic Agents - 317059 Adefovir Acamprosate 8:18.32 - Nucleosides and Nucleotides - 302036 28:92 - Central Nervous System Agents, Adenosine 24:04.04.24 - Class IV Antiarrhythmics - 304010 Acarbose Adenovirus Vaccine Live Oral 68:20.02 - alpha-Glucosidase Inhibitors - 396015 80:12 - Vaccines - 315016 Acebutolol Ado-Trastuzumab 24:24 - beta-Adrenergic Blocking Agents - 387003 10:00 - Antineoplastic Agents - 313041 12:16.08.08 - Selective -

Keeping up with FDA Drug Approvals: 60 New Drugs in 60 Minutes Elizabeth A
Keeping Up with FDA Drug Approvals: 60 New Drugs in 60 Minutes Elizabeth A. Shlom, PharmD, BCPS Senior Vice President & Director Clinical Pharmacy Program | Acurity, Inc. Privileged and Confidential April 10, 2019 Privileged and Confidential Program Objectives By the end of the presentation, the pharmacist or pharmacy technician participant will be able to: ▪ Identify orphan drugs and first-in-class medications approved by the FDA in 2018. ▪ List five new drugs and their indications. ▪ Identify the place in therapy for three novel monoclonal antibodies. ▪ Discuss at least two new medications that address public health concerns. Dr. Shlom does not have any conflicts of interest in regard to this presentation. Both trade names and generic names will be discussed throughout the presentation Privileged and Confidential 2018 NDA Approvals (NMEs/BLAs) ▪ Lutathera (lutetium Lu 177 dotatate) ▪ Braftovi (encorafenib) ▪ Vizimpro (dacomitinib) ▪ Biktarvy (bictegravir, emtricitabine, ▪ TPOXX (tecovirimat) ▪ Libtayo (cemiplimab-rwic) tenofovir, ▪ Tibsovo (ivosidenib) ▪ Seysara (sarecycline) alafenamide) ▪ Krintafel (tafenoquine) ▪ Nuzyra (omadacycline) ▪ Symdeko (tezacaftor, ivacaftor) ▪ Orilissa (elagolix sodium) ▪ Revcovi (elapegademase-lvir) ▪ Erleada (apalutamide) ▪ Omegaven (fish oil triglycerides) ▪ Tegsedi (inotersen) ▪ Trogarzo (ibalizumab-uiyk) ▪ Mulpleta (lusutrombopag) ▪ Talzenna (talazoparib) ▪ Ilumya (tildrakizumab-asmn) ▪ Poteligeo (mogamulizumab-kpkc) ▪ Xofluza (baloxavir marboxil) ▪ Tavalisse (fostamatinib disodium) ▪ Onpattro (patisiran) -

The Role of Brincidofovir in Preparation for a Potential Smallpox Outbreak
viruses Perspective The Role of Brincidofovir in Preparation for a Potential Smallpox Outbreak Scott A. Foster 1,*, Scott Parker 2 ID and Randall Lanier 1 1 Chimerix, Durham, NC 27713, USA; [email protected] 2 Department of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, St. Louis, MO 63104, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-919-597-7741 Received: 29 September 2017; Accepted: 26 October 2017; Published: 30 October 2017 Abstract: Smallpox (variola) virus is considered a Category A bioterrorism agent due to its ability to spread rapidly and the high morbidity and mortality rates associated with infection. Current recommendations recognize the importance of oral antivirals and call for having at least two smallpox antivirals with different mechanisms of action available in the event of a smallpox outbreak. Multiple antivirals are recommended due in large part to the propensity of viruses to become resistant to antiviral therapy, especially monotherapy. Advances in synthetic biology heighten concerns that a bioterror attack with variola would utilize engineered resistance to antivirals and potentially vaccines. Brincidofovir, an oral antiviral in late stage development, has proven effective against orthopoxviruses in vitro and in vivo, has a different mechanism of action from tecovirimat (the only oral smallpox antiviral currently in the US Strategic National Stockpile), and has a resistance profile that reduces concerns in the scenario of a bioterror attack using genetically engineered smallpox. Given the devastating potential of smallpox as a bioweapon, preparation of a multi-pronged defense that accounts for the most obvious bioengineering possibilities is strategically imperative. Keywords: smallpox; variola virus; bioterrorism; bioweapon; brincidofovir; CMX001; antiviral 1. -

2019 Icar Program & Abstracts Book
Hosted by the International Society for Antiviral Research (ISAR) ND International Conference 32on Antiviral Research (ICAR) Baltimore MARYLAND PROGRAM and USA Hyatt Regency BALTIMORE ABSTRACTS May 12-15 2019 ND TABLE OF International Conference CONTENTS 32on Antiviral Research (ICAR) Daily Schedule . .3 Organization . 4 Contributors . 5 Keynotes & Networking . 6 Schedule at a Glance . 7 ISAR Awardees . 10 The 2019 Chu Family Foundation Scholarship Awardees . 15 Speaker Biographies . 17 Program Schedule . .25 Posters . 37 Abstracts . 53 Author Index . 130 PROGRAM and ABSTRACTS of the 32nd International Conference on Antiviral Research (ICAR) 2 ND DAILY International Conference SCHEDULE 32on Antiviral Research (ICAR) SUNDAY, MAY 12, 2019 › Women in Science Roundtable › Welcome and Keynote Lectures › Antonín Holý Memorial Award Lecture › Influenza Symposium › Opening Reception MONDAY, MAY 13, 2019 › Women in Science Award Lecture › Emerging Virus Symposium › Short Presentations 1 › Poster Session 1 › Retrovirus Symposium › ISAR Award of Excellence Presentation › PechaKucha Event with Introduction of First Time Attendees TUESDAY, MAY 14, 2019 › What’s New in Antiviral Research 1 › Short Presentations 2 & 3 › ISAR Award for Outstanding Contributions to the Society Presentation › Career Development Panel › William Prusoff Young Investigator Award Lecture › Medicinal Chemistry Symposium › Poster Session 2 › Networking Reception WEDNESDAY, MAY 15, 2019 › Gertrude Elion Memorial Award Lecture › What’s New in Antiviral Research 2 › Shotgun Oral -

Synthesis of Polycyclic Compounds with Antiviral Activity
Synthesis of polycyclic compounds with antiviral activity Eva Torres Costa ADVERTIMENT. La consulta d’aquesta tesi queda condicionada a l’acceptació de les següents condicions d'ús: La difusió d’aquesta tesi per mitjà del servei TDX (www.tdx.cat) i a través del Dipòsit Digital de la UB (diposit.ub.edu) ha estat autoritzada pels titulars dels drets de propietat intel·lectual únicament per a usos privats emmarcats en activitats d’investigació i docència. No s’autoritza la seva reproducció amb finalitats de lucre ni la seva difusió i posada a disposició des d’un lloc aliè al servei TDX ni al Dipòsit Digital de la UB. No s’autoritza la presentació del seu contingut en una finestra o marc aliè a TDX o al Dipòsit Digital de la UB (framing). Aquesta reserva de drets afecta tant al resum de presentació de la tesi com als seus continguts. En la utilització o cita de parts de la tesi és obligat indicar el nom de la persona autora. ADVERTENCIA. La consulta de esta tesis queda condicionada a la aceptación de las siguientes condiciones de uso: La difusión de esta tesis por medio del servicio TDR (www.tdx.cat) y a través del Repositorio Digital de la UB (diposit.ub.edu) ha sido autorizada por los titulares de los derechos de propiedad intelectual únicamente para usos privados enmarcados en actividades de investigación y docencia. No se autoriza su reproducción con finalidades de lucro ni su difusión y puesta a disposición desde un sitio ajeno al servicio TDR o al Repositorio Digital de la UB. -

Drug Pipeline Monthly Update
Drug Pipeline Monthly Update Critical updates in an ever changing environment December 2018 New drug information ● Temixys™ (lamivudine/tenofovir disoproxil fumarate): The Food and Drug Administration (FDA) approved Celltrion’s Temixys in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 35 kg, dosed once daily. Temixys is a combination of two nucleoside reverse transcriptase inhibitors (NRTIs) in one tablet. Celltrion intends to sell Temixys at a discounted price compared to Gilead’s Truvada® (emtricitabine/tenofovir disoproxil fumarate).1 Celltrion plans to launch early 2019.1 ● Tolsura™ (itraconazole) capsules: Mayne Pharma received FDA approval for Tolsura for the treatment of systemic fungal infections in adult patients including certain forms of: → blastomycosis, → histoplasmosis, and → aspergillosis Mayne notes that this is a Super-BioAvailable (SUBA) formulation of itraconazole that has been shown in clinical studies to have increased bioavailability and reduced variability when compared to other itraconazole formulations.2 Tolsura will launch in January 2019.2 ● Montegrity™ (prucalopride): The FDA approved Shire’s Montegrity, a once daily, oral medication for the treatment of adults with chronic idiopathic constipation (CIC). Montegrity will compete for market share in the CIC space with Allergan’s Linzess® (linaclotide), Takeda’s Amitiza® (lubiprostone), and Synergy’s Trulance® (plecanatide). Montegrity is anticipated to launch in 2019. continued While the information in this newsletter is from sources we believe to be reliable, we do not warrant that the information in this document is free from error. Use it only as a guide. Statements regarding drugs or manufacturers are not intended as promotion; those statements should not be used to make assumptions about formulary status. -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

Research on Substances with Activity Against Orthopoxviruses
Annals of Agricultural and Environmental Medicine 2013, Vol 20, No 1, 1-7 REVIEW ARTICLE www.aaem.pl Research on substances with activity against orthopoxviruses Marcin Kołodziej1, Justyna Joniec1, Michał Bartoszcze1, Romuald Gryko1, Janusz Kocik2, Józef Knap3 1 Biological Threats Identification and Countermeasure Center, Military Institute of Hygiene and Epidemiology, Puławy, Poland 2 Military Institute of Hygiene and Epidemiology, Warsaw, Poland 3 Department of Epidemiology, Medical University, Warsaw, Poland Kołodziej M, Joniec J, Bartoszcze M, Gryko R, Kocik J, Knap J. Research on substances with activity against orthopoxviruses. Ann Agric Environ Med. 2013; 20(1): 1-7. Abstract Although smallpox was eradicated over 30 years ago, the disease remains a major threat. High mortality, high infectivity and low resistance of the contemporary population make the smallpox virus very attractive to terrorists. The possible presence of illegal stocks of the virus or risk of deliberate genetic modifications cause serious concerns among experts. Hence, it is reasonable to seek effective drugs that could be used in case of smallpox outbreak. This paper reviews studies on compounds with proven in vitro or in vivo antipoxviruses potential, which show various mechanisms of action. Nucleoside analogues, such as cidofovir, can inhibit virus replication. Cidofovir derivatives are developed to improve the bioavailability of the drug. Among the nucleoside analogues under current investigation are: ANO (adenozine N1-oxide) and its derivatives, N-methanocarbothymidine [(N)-MCT], or derivatitives of aciklovir, peninclovir and brivudin. Recently, ST-246 – which effectively inhibits infection by limiting release of progeny virions – has become an object of attention. It has been also been demonstrated that compounds such as: nigericin, aptamers and peptides may have antiviral potential. -

TPOXX (Tecovirimat) Capsules for Oral Use None (4) Initial U.S
HIGHLIGHTS OF PRESCRIBING INFORMATION ______________ ______________ These highlights do not include all the information needed to use DOSAGE FORMS AND STRENGTHS TPOXX® safely and effectively. See full prescribing information for Capsule: 200 mg. (3) TPOXX. ___________________ ____________________ CONTRAINDICATIONS TPOXX (tecovirimat) capsules for oral use None (4) Initial U.S. Approval: 2018 _______________ _______________ WARNINGS AND PRECAUTIONS __________________ _________________ INDICATIONS AND USAGE Hypoglycemia: Co-administration with repaglinide may cause hypoglycemia. TPOXX is an inhibitor of the orthopoxvirus VP37 envelope wrapping protein Monitor blood glucose and monitor for hypoglycemic symptoms during co- and is indicated for the treatment of human smallpox disease in adults and administration. (5.1) pediatric patients weighing at least 13 kg. (1.1) ____________________ ____________________ Limitations of Use: ADVERSE REACTIONS The effectiveness of TPOXX for treatment of smallpox disease has not Common adverse reactions in healthy adult subjects (≥ 2%) were headache, been determined in humans because adequate and well-controlled field nausea, abdominal pain, and vomiting. (6.1) trials have not been feasible, and inducing smallpox disease in humans to study the drug’s efficacy is not ethical. (1.2) To report SUSPECTED ADVERSE REACTIONS, contact SIGA TPOXX efficacy may be reduced in immunocompromised patients based Technologies at 1-541-753-2000 or FDA at 1-800-FDA-1088 or on studies demonstrating reduced efficacy in immunocompromised animal www.fda.gov/medwatch. models. (1.2) ____________________ ____________________ DRUG INTERACTIONS _______________ ______________ DOSAGE AND ADMINISTRATION Consult the full prescribing information prior to and during treatment for TPOXX should be taken within 30 minutes after a full meal of moderate or potential drug interactions. -

A Computational Approach to Aid Clinicians in Selecting Anti-Viral
www.nature.com/scientificreports OPEN A computational approach to aid clinicians in selecting anti‑viral drugs for COVID‑19 trials Aanchal Mongia1, Sanjay Kr. Saha2, Emilie Chouzenoux3* & Angshul Majumdar1* The year 2020 witnessed a heavy death toll due to COVID‑19, calling for a global emergency. The continuous ongoing research and clinical trials paved the way for vaccines. But, the vaccine efcacy in the long run is still questionable due to the mutating coronavirus, which makes drug re‑positioning a reasonable alternative. COVID‑19 has hence fast‑paced drug re‑positioning for the treatment of COVID‑19 and its symptoms. This work builds computational models using matrix completion techniques to predict drug‑virus association for drug re‑positioning. The aim is to assist clinicians with a tool for selecting prospective antiviral treatments. Since the virus is known to mutate fast, the tool is likely to help clinicians in selecting the right set of antivirals for the mutated isolate. The main contribution of this work is a manually curated database publicly shared, comprising of existing associations between viruses and their corresponding antivirals. The database gathers similarity information using the chemical structure of drugs and the genomic structure of viruses. Along with this database, we make available a set of state‑of‑the‑art computational drug re‑positioning tools based on matrix completion. The tools are frst analysed on a standard set of experimental protocols for drug target interactions. The best performing ones are applied for the task of re‑positioning antivirals for COVID‑19. These tools select six drugs out of which four are currently under various stages of trial, namely Remdesivir (as a cure), Ribavarin (in combination with others for cure), Umifenovir (as a prophylactic and cure) and Sofosbuvir (as a cure).