A Computational Approach to Aid Clinicians in Selecting Anti-Viral
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Development of Antiviral Agents for Enteroviruses
Journal of Antimicrobial Chemotherapy (2008) 62, 1169–1173 doi:10.1093/jac/dkn424 Advance Access publication 18 October 2008 Development of antiviral agents for enteroviruses Tzu-Chun Chen1–3, Kuo-Feng Weng1,2, Shih-Cheng Chang1,2, Jing-Yi Lin1,2, Peng-Nien Huang1,2 and Shin-Ru Shih1,2,4,5* 1Research Center for Emerging Viral Infections, Chang Gung University, Taoyuan, Taiwan; 2Department of Medical Biotechnology and Laboratory Science, Chang Gung University, Taoyuan, Taiwan; 3Institute Downloaded from https://academic.oup.com/jac/article/62/6/1169/774341 by guest on 26 September 2021 of Molecular Medicine, National Tsing Hua University, Hsinchu, Taiwan; 4Clinical Virology Laboratory, Chang Gung Memorial Hospital, Taoyuan, Taiwan; 5Division of Biotechnology and Pharmaceutical Research, National Health Research Institutes, Chunan, Taiwan Enteroviruses (EVs) are common human pathogens that are associated with numerous disease symptoms in many organ systems of the body. Although EV infections commonly cause mild or non- symptomatic illness, some of them are associated with severe diseases such as CNS complications. The current absence of effective vaccines for most viral infection and no available antiviral drugs for the treatment of EVs highlight the urgency and significance of developing antiviral agents. Several key steps in the viral life cycle are potential targets for blocking viral replication. This article reviews recent studies of antiviral developments for EVs based on various molecular targets that interrupt viral attach- ment, viral translation, polyprotein processing and RNA replication. Keywords: capsid proteins, viral proteases, viral RNA replication, 50 untranslated region Introduction EVs such as echovirus 6, 9 and 30 and CVB5 were the most common causes of aseptic meningitis in children.4 EV70 and Enteroviruses (EVs) are a common cause of infections in CVA24 were associated with acute haemorrhagic conjunctivitis.4 humans, especially children. -

COVID-19) Pandemic on National Antimicrobial Consumption in Jordan
antibiotics Article An Assessment of the Impact of Coronavirus Disease (COVID-19) Pandemic on National Antimicrobial Consumption in Jordan Sayer Al-Azzam 1, Nizar Mahmoud Mhaidat 1, Hayaa A. Banat 2, Mohammad Alfaour 2, Dana Samih Ahmad 2, Arno Muller 3, Adi Al-Nuseirat 4 , Elizabeth A. Lattyak 5, Barbara R. Conway 6,7 and Mamoon A. Aldeyab 6,* 1 Clinical Pharmacy Department, Jordan University of Science and Technology, Irbid 22110, Jordan; [email protected] (S.A.-A.); [email protected] (N.M.M.) 2 Jordan Food and Drug Administration (JFDA), Amman 11181, Jordan; [email protected] (H.A.B.); [email protected] (M.A.); [email protected] (D.S.A.) 3 Antimicrobial Resistance Division, World Health Organization, Avenue Appia 20, 1211 Geneva, Switzerland; [email protected] 4 World Health Organization Regional Office for the Eastern Mediterranean, Cairo 11371, Egypt; [email protected] 5 Scientific Computing Associates Corp., River Forest, IL 60305, USA; [email protected] 6 Department of Pharmacy, School of Applied Sciences, University of Huddersfield, Huddersfield HD1 3DH, UK; [email protected] 7 Institute of Skin Integrity and Infection Prevention, University of Huddersfield, Huddersfield HD1 3DH, UK * Correspondence: [email protected] Citation: Al-Azzam, S.; Mhaidat, N.M.; Banat, H.A.; Alfaour, M.; Abstract: Coronavirus disease 2019 (COVID-19) has overlapping clinical characteristics with bacterial Ahmad, D.S.; Muller, A.; Al-Nuseirat, respiratory tract infection, leading to the prescription of potentially unnecessary antibiotics. This A.; Lattyak, E.A.; Conway, B.R.; study aimed at measuring changes and patterns of national antimicrobial use for one year preceding Aldeyab, M.A. -

The Protean Neurologic Manifestations of Varicella-Zoster Virus Infection
REVIEW MARIA A. NAGEL, MD DONALD H. GILDEN, MD Department of Neurology, University of Departments of Neurology and Microbiology, Colorado Health Sciences Center, Denver University of Colorado Health Sciences Center, Denver The protean neurologic manifestations of varicella-zoster virus infection ■ ABSTRACT ARICELLA-ZOSTER VIRUS (VZV) is an V exclusively human, highly neurotropic Multiple neurologic complications may follow the alphaherpesvirus. Primary infection causes reactivation of varicella-zoster virus (VZV), including chickenpox, after which the virus becomes herpes zoster (also known as zoster or shingles), latent in cranial nerve ganglia, dorsal root postherpetic neuralgia, vasculopathy, myelitis, necrotizing ganglia, and autonomic ganglia along the retinitis, and zoster sine herpete (pain without rash). entire neuraxis. Decades later, VZV can reac- These conditions can be difficult to recognize, especially tivate to cause a number of neurologic condi- as several can occur without rash. tions. This article discusses the protean manifes- ■ KEY POINTS tations of VZV reactivation and their diagno- sis and treatment. The most common sequela of herpes zoster is postherpetic neuralgia, which can persist for months and ■ VZV REACTIVATION sometimes years after the rash resolves. HAS MANY MANIFESTATIONS VZV vasculopathy can present as transient ischemic VZV reactivation can produce a number of neurologic complications: attacks, ischemic or hemorrhagic stroke, or aneurysm. •Herpes zoster (shingles), manifesting as pain and rash in up to three dermatomes Vasculopathy and neurologic complications of VZV are • Postherpetic neuralgia, the most common best diagnosed by detecting VZV DNA in bodily fluids or sequela of zoster, is pain that persists for tissues or anti-VZV immunoglobulin G in the months and sometimes years after the rash cerebrospinal fluid. -

Dectova, INN-Zanamivir
28 February 2019 EMA/CHMP/851480/2018 Committee for Medicinal Products for Human Use (CHMP) Assessment report DECTOVA International non-proprietary name: zanamivir Procedure No. EMEA/H/C/004102/0000 Note Assessment report as adopted by the CHMP with all information of a commercially confidential nature deleted. Official address Domenico Scarlattilaan 6 ● 1083 HS Amsterdam ● The Netherlands Address for visits and deliveries Refer to www.ema.europa.eu/how-to-find-us Send us a question Go to www.ema.europa.eu/contact Telephone +31 (0)88 781 6000 An agency of the European Union © European Medicines Agency, 2019. Reproduction is authorised provided the source is acknowledged. Table of contents 1. Background information on the procedure .............................................. 7 1.1. Submission of the dossier ...................................................................................... 7 1.2. Steps taken for the assessment of the product ......................................................... 9 2. Scientific discussion .............................................................................. 12 2.1. Problem statement ............................................................................................. 12 2.1.1. Disease or condition ......................................................................................... 12 2.1.2. Epidemiology .................................................................................................. 12 2.1.3. Biologic features ............................................................................................. -

(MGH) COVID-19 Treatment Guidance
Version 8.0 4/28/2021 10:00AM © Copyright 2020 The General Hospital Corporation. All Rights Reserved. Massachusetts General Hospital (MGH) COVID-19 Treatment Guidance This document was prepared (in March, 2020-April, 2021) by and for MGH medical professionals (a.k.a. clinicians, care givers) and is being made available publicly for informational purposes only, in the context of a public health emergency related to COVID-19 (a.k.a. the coronavirus) and in connection with the state of emergency declared by the Governor of the Commonwealth of Massachusetts and the President of the United States. It is neither an attempt to substitute for the practice of medicine nor as a substitute for the provision of any medical professional services. Furthermore, the content is not meant to be complete, exhaustive, or a substitute for medical professional advice, diagnosis, or treatment. The information herein should be adapted to each specific patient based on the treating medical professional’s independent professional judgment and consideration of the patient’s needs, the resources available at the location from where the medical professional services are being provided (e.g., healthcare institution, ambulatory clinic, physician’s office, etc.), and any other unique circumstances. This information should not be used to replace, substitute for, or overrule a qualified medical professional’s judgment. This website may contain third party materials and/or links to third party materials and third party websites for your information and convenience. Partners is not responsible for the availability, accuracy, or content of any of those third party materials or websites nor does it endorse them. -

Infectious Diseases
2013 MEDICINES IN DEVELOPMENT REPORT Infectious Diseases A Report on Diseases Caused by Bacteria, Viruses, Fungi and Parasites PRESENTED BY AMERICA’S BIOPHARMACEUTICAL RESEARCH COMPANIES Biopharmaceutical Research Evolves Against Infectious Diseases with Nearly 400 Medicines and Vaccines in Testing Throughout history, infectious diseases hepatitis C that inhibits the enzyme have taken a devastating toll on the lives essential for viral replication. and well-being of people around the • An anti-malarial drug that has shown Medicines in Development world. Caused when pathogens such activity against Plasmodium falci- For Infectious Diseases as bacteria or viruses enter a body and parum malaria which is resistant to multiply, infectious diseases were the current treatments. Application leading cause of death in the United Submitted States until the 1920s. Today, vaccines • A potential new antibiotic to treat methicillin-resistant Staphylococcus Phase III and infectious disease treatments have proven to be effective treatments in aureus (MRSA). Phase II many cases, but infectious diseases still • A novel treatment that works by Phase I pose a very serious threat to patients. blocking the ability of the smallpox Recently, some infectious pathogens, virus to spread to other cells, thus 226 such as pseudomonas bacteria, have preventing it from causing disease. become resistant to available treatments. Infectious diseases may never be fully Diseases once considered conquered, eradicated. However, new knowledge, such as tuberculosis, have reemerged new technologies, and the continuing as a growing health threat. commitment of America’s biopharma- America’s biopharmaceutical research ceutical research companies can help companies are developing 394 medicines meet the continuing—and ever-changing and vaccines to combat the many threats —threat from infectious diseases. -

Current Therapies for Chronic Hepatitis C
Southern Illinois University Edwardsville SPARK Pharmacy Faculty Research, Scholarship, and Creative Activity School of Pharmacy 1-2011 Current Therapies for Chronic Hepatitis C McKenzie C. Ferguson Southern Illinois University Edwardsville, [email protected] Follow this and additional works at: https://spark.siue.edu/pharmacy_fac Part of the Pharmacy and Pharmaceutical Sciences Commons Recommended Citation Ferguson, McKenzie C., "Current Therapies for Chronic Hepatitis C" (2011). Pharmacy Faculty Research, Scholarship, and Creative Activity. 4. https://spark.siue.edu/pharmacy_fac/4 This Article is brought to you for free and open access by the School of Pharmacy at SPARK. It has been accepted for inclusion in Pharmacy Faculty Research, Scholarship, and Creative Activity by an authorized administrator of SPARK. For more information, please contact [email protected],[email protected]. Chronic Hepatitis C & Current Therapies 1 Review of Chronic Hepatitis C & Current Therapies Reviews of Therapeutics Pharmacotherapy McKenzie C. Ferguson, Pharm.D., BCPS From the School of Pharmacy, Southern Illinois University Edwardsville, Department of Pharmacy Practice, Edwardsville, Illinois. Address for reprint requests : Southern Illinois University Edwardsville School of Pharmacy 220 University Park Drive, Ste 1037 Edwardsville, IL 62026-2000 Email: [email protected] Keywords : hepatitis C, HCV, ribavirin, interferon, peginterferon alfa-2a, peginterferon alfa-2b, albinterferon, taribavirin, telaprevir, therapeutic efficacy, safety The author received no sources of support in the form of grants, equipment, or drugs. Chronic Hepatitis C & Current Therapies 2 ABSTRACT Hepatitis C virus affects more than 180 million people worldwide and as many as 4 million people in the United States. Given that most patients are asymptomatic until late in disease progression, diagnostic screening and evaluation of patients that display high-risk behaviors associated with acquisition of hepatitis C should be performed. -
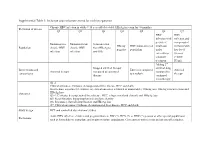
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

The Evolution of Pleconaril: Modified O-Alkyl Linker Analogs Have
molecules Communication The Evolution of Pleconaril: Modified O-Alkyl Linker Analogs Have Biological Activity towards Coxsackievirus B3 Nancy 1, 2, 1 3 Alexandrina Volobueva y, Anna Egorova y, Anastasia Galochkina , Sean Ekins , Vladimir Zarubaev 1 and Vadim Makarov 2,* 1 Saint-Petersburg Pasteur Institute, Mira str., 14, 197101 Saint Petersburg, Russia; [email protected] (A.V.); [email protected] (A.G.); [email protected] (V.Z.) 2 Bach Institute of Biochemistry, Research Center of Biotechnology of the Russian Academy of Sciences, Leninsky prospect, 33, build. 2, 119071 Moscow, Russia; [email protected] 3 Collaborations Pharmaceuticals, Inc., 840 Main Campus Drive, Lab 3510, Raleigh, NC 27606, USA; [email protected] * Correspondence: [email protected] These authors contributed equally to this work. y Received: 10 February 2020; Accepted: 13 March 2020; Published: 16 March 2020 Abstract: Coxsackieviruses type B are one of the most common causes of mild upper respiratory and gastrointestinal illnesses. At the time of writing, there are no approved drugs for effective antiviral treatment for Coxsackieviruses type B. We used the core-structure of pleconaril, a well-known antienteroviral drug candidate, for the synthesis of novel compounds with O-propyl linker modifications. Some original compounds with 4 different linker patterns, such as sulfur atom, ester, amide, and piperazine, were synthesized according to five synthetic schemes. The cytotoxicity and bioactivity of 14 target compounds towards Coxsackievirus B3 Nancy were examined. Based on the results, the values of 50% cytotoxic dose (CC50), 50% virus-inhibiting dose (IC50), and selectivity index (SI) were calculated for each compound. Several of the novel synthesized derivatives exhibited a strong anti-CVB3 activity (SI > 20 to > 200). -

Antiviral Agents Active Against Influenza a Viruses
REVIEWS Antiviral agents active against influenza A viruses Erik De Clercq Abstract | The recent outbreaks of avian influenza A (H5N1) virus, its expanding geographic distribution and its ability to transfer to humans and cause severe infection have raised serious concerns about the measures available to control an avian or human pandemic of influenza A. In anticipation of such a pandemic, several preventive and therapeutic strategies have been proposed, including the stockpiling of antiviral drugs, in particular the neuraminidase inhibitors oseltamivir (Tamiflu; Roche) and zanamivir (Relenza; GlaxoSmithKline). This article reviews agents that have been shown to have activity against influenza A viruses and discusses their therapeutic potential, and also describes emerging strategies for targeting these viruses. HXNY In the face of the persistent threat of human influenza A into the interior of the virus particles (virions) within In the naming system for (H3N2, H1N1) and B infections, the outbreaks of avian endosomes, a process that is needed for the uncoating virus strains, H refers to influenza (H5N1) in Southeast Asia, and the potential of to occur. The H+ ions are imported through the M2 haemagglutinin and N a new human or avian influenza A variant to unleash a (matrix 2) channels10; the transmembrane domain of to neuraminidase. pandemic, there is much concern about the shortage in the M2 protein, with the amino-acid residues facing both the number and supply of effective anti-influenza- the ion-conducting pore, is shown in FIG. 3a (REF. 11). virus agents1–4. There are, in principle, two mechanisms Amantadine has been postulated to block the interior by which pandemic influenza could originate: first, by channel within the tetrameric M2 helix bundle12. -

CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene and Pathophysiological Conditions
Review CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene and Pathophysiological Conditions Rossana Roncato 1,*,†, Jacopo Angelini 2,†, Arianna Pani 2,3,†, Erika Cecchin 1, Andrea Sartore-Bianchi 2,4, Salvatore Siena 2,4, Elena De Mattia 1, Francesco Scaglione 2,3,‡ and Giuseppe Toffoli 1,‡ 1 1 Experimental and Clinical Pharmacology Unit, Centro di Riferimento Oncologico (CRO), IRCCS, 33081 Aviano, Italy; [email protected] (E.C.); [email protected] (E.D.M.); [email protected] (G.T.) 2 Department of Oncology and Hemato-Oncology, Università degli Studi di Milano, 20122 Milan, Italy; [email protected] (J.A.); [email protected] (A.P.); [email protected] (A.S-B.); [email protected] (S.S.); [email protected] (F.S.) 3 Clinical Pharmacology Unit, ASST Grande Ospedale Metropolitano Niguarda, Piazza dell'Ospedale Maggiore 3, 20162 Milan, Italy 4 Department of Hematology and Oncology, Niguarda Cancer Center, Grande Ospedale Metropolitano Niguarda, 20162 Milan, Italy * Correspondence: [email protected]; Tel.:+390434659130 † These authors contributed equally. ‡ These authors share senior authorship. Int. J. Mol. Sci. 2020, 21, x; doi: www.mdpi.com/journal/ijms Int. J. Mol. Sci. 2020, 21, x 2 of 8 Table S1. Co-administered agents categorized according to their potential risk for Drug-Drug interaction (DDI) in combination with CDK4/6 inhibitors (CDKis). Colors suggest the risk of DDI with CDKis: green, low risk DDI; orange, moderate risk DDI; red, high risk DDI. ADME, absorption, distribution, metabolism, and excretion; GI, Gastrointestinal; TdP, Torsades de Pointes; NTI, narrow therapeutic index. * Cardiological toxicity should be considered especially for ribociclib due to the QT prolongation. -

2020 Taxonomic Update for Phylum Negarnaviricota (Riboviria: Orthornavirae), Including the Large Orders Bunyavirales and Mononegavirales
Archives of Virology https://doi.org/10.1007/s00705-020-04731-2 VIROLOGY DIVISION NEWS 2020 taxonomic update for phylum Negarnaviricota (Riboviria: Orthornavirae), including the large orders Bunyavirales and Mononegavirales Jens H. Kuhn1 · Scott Adkins2 · Daniela Alioto3 · Sergey V. Alkhovsky4 · Gaya K. Amarasinghe5 · Simon J. Anthony6,7 · Tatjana Avšič‑Županc8 · María A. Ayllón9,10 · Justin Bahl11 · Anne Balkema‑Buschmann12 · Matthew J. Ballinger13 · Tomáš Bartonička14 · Christopher Basler15 · Sina Bavari16 · Martin Beer17 · Dennis A. Bente18 · Éric Bergeron19 · Brian H. Bird20 · Carol Blair21 · Kim R. Blasdell22 · Steven B. Bradfute23 · Rachel Breyta24 · Thomas Briese25 · Paul A. Brown26 · Ursula J. Buchholz27 · Michael J. Buchmeier28 · Alexander Bukreyev18,29 · Felicity Burt30 · Nihal Buzkan31 · Charles H. Calisher32 · Mengji Cao33,34 · Inmaculada Casas35 · John Chamberlain36 · Kartik Chandran37 · Rémi N. Charrel38 · Biao Chen39 · Michela Chiumenti40 · Il‑Ryong Choi41 · J. Christopher S. Clegg42 · Ian Crozier43 · John V. da Graça44 · Elena Dal Bó45 · Alberto M. R. Dávila46 · Juan Carlos de la Torre47 · Xavier de Lamballerie38 · Rik L. de Swart48 · Patrick L. Di Bello49 · Nicholas Di Paola50 · Francesco Di Serio40 · Ralf G. Dietzgen51 · Michele Digiaro52 · Valerian V. Dolja53 · Olga Dolnik54 · Michael A. Drebot55 · Jan Felix Drexler56 · Ralf Dürrwald57 · Lucie Dufkova58 · William G. Dundon59 · W. Paul Duprex60 · John M. Dye50 · Andrew J. Easton61 · Hideki Ebihara62 · Toufc Elbeaino63 · Koray Ergünay64 · Jorlan Fernandes195 · Anthony R. Fooks65 · Pierre B. H. Formenty66 · Leonie F. Forth17 · Ron A. M. Fouchier48 · Juliana Freitas‑Astúa67 · Selma Gago‑Zachert68,69 · George Fú Gāo70 · María Laura García71 · Adolfo García‑Sastre72 · Aura R. Garrison50 · Aiah Gbakima73 · Tracey Goldstein74 · Jean‑Paul J. Gonzalez75,76 · Anthony Grifths77 · Martin H. Groschup12 · Stephan Günther78 · Alexandro Guterres195 · Roy A.