The Role of Brincidofovir in Preparation for a Potential Smallpox Outbreak
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Multicenter, Multi-Outbreak, Randomized, Controlled Safety And
2018 Ebola MCM RCT Protocol Page 1 of 92 IND#: 125530 CONFIDENTIAL 4 October 2019, version 7.0 A Multicenter, Multi-Outbreak, Randomized, Controlled Safety and Efficacy Study of Investigational Therapeutics for the Treatment of Patients with Ebola Virus Disease Short Title: 2018 Ebola MCM RCT Protocol Sponsored by: Office of Clinical Research Policy and Regulatory Operations (OCRPRO) National Institute of Allergy and Infectious Diseases 5601 Fishers Lane Bethesda, MD 20892 NIH Protocol Number: 19-I-0003 Protocol IND #: 125530 ClinicalTrials.gov Number: NCT03719586 Date: 4 October 2019 Version: 7.0 CONFIDENTIAL This document is confidential. No part of it may be transmitted, reproduced, published, or used by other persons without prior written authorization from the study sponsor and principal investigator. 2018 Ebola MCM RCT Protocol Page 2 of 92 IND#: 125530 CONFIDENTIAL 4 October 2019, version 7.0 KEY ROLES DRC Principal Investigator: Jean-Jacques Muyembe-Tamfum, MD, PhD Director-General, DRC National Institute for Biomedical Research Professor of Microbiology, Kinshasa University Medical School Kinshasa Gombe Democratic Republic of the Congo Phone: +243 898949289 Email: [email protected] Other International Investigators: see Appendix E Statistical Lead: Lori Dodd, PhD Biostatistics Research Branch, DCR, NIAID 5601 Fishers Lane, Room 4C31 Rockville, MD 20852 Phone: 240-669-5247 Email: [email protected] U.S. Principal Investigator: Richard T. Davey, Jr., MD Clinical Research Section, LIR, NIAID, NIH Building 10, Room 4-1479, Bethesda, -

Infectious Diseases
2013 MEDICINES IN DEVELOPMENT REPORT Infectious Diseases A Report on Diseases Caused by Bacteria, Viruses, Fungi and Parasites PRESENTED BY AMERICA’S BIOPHARMACEUTICAL RESEARCH COMPANIES Biopharmaceutical Research Evolves Against Infectious Diseases with Nearly 400 Medicines and Vaccines in Testing Throughout history, infectious diseases hepatitis C that inhibits the enzyme have taken a devastating toll on the lives essential for viral replication. and well-being of people around the • An anti-malarial drug that has shown Medicines in Development world. Caused when pathogens such activity against Plasmodium falci- For Infectious Diseases as bacteria or viruses enter a body and parum malaria which is resistant to multiply, infectious diseases were the current treatments. Application leading cause of death in the United Submitted States until the 1920s. Today, vaccines • A potential new antibiotic to treat methicillin-resistant Staphylococcus Phase III and infectious disease treatments have proven to be effective treatments in aureus (MRSA). Phase II many cases, but infectious diseases still • A novel treatment that works by Phase I pose a very serious threat to patients. blocking the ability of the smallpox Recently, some infectious pathogens, virus to spread to other cells, thus 226 such as pseudomonas bacteria, have preventing it from causing disease. become resistant to available treatments. Infectious diseases may never be fully Diseases once considered conquered, eradicated. However, new knowledge, such as tuberculosis, have reemerged new technologies, and the continuing as a growing health threat. commitment of America’s biopharma- America’s biopharmaceutical research ceutical research companies can help companies are developing 394 medicines meet the continuing—and ever-changing and vaccines to combat the many threats —threat from infectious diseases. -
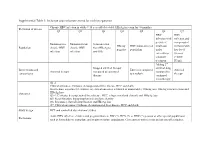
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

AHFS Pharmacologic-Therapeutic Classification System
AHFS Pharmacologic-Therapeutic Classification System Abacavir 48:24 - Mucolytic Agents - 382638 8:18.08.20 - HIV Nucleoside and Nucleotide Reverse Acitretin 84:92 - Skin and Mucous Membrane Agents, Abaloparatide 68:24.08 - Parathyroid Agents - 317036 Aclidinium Abatacept 12:08.08 - Antimuscarinics/Antispasmodics - 313022 92:36 - Disease-modifying Antirheumatic Drugs - Acrivastine 92:20 - Immunomodulatory Agents - 306003 4:08 - Second Generation Antihistamines - 394040 Abciximab 48:04.08 - Second Generation Antihistamines - 394040 20:12.18 - Platelet-aggregation Inhibitors - 395014 Acyclovir Abemaciclib 8:18.32 - Nucleosides and Nucleotides - 381045 10:00 - Antineoplastic Agents - 317058 84:04.06 - Antivirals - 381036 Abiraterone Adalimumab; -adaz 10:00 - Antineoplastic Agents - 311027 92:36 - Disease-modifying Antirheumatic Drugs - AbobotulinumtoxinA 56:92 - GI Drugs, Miscellaneous - 302046 92:20 - Immunomodulatory Agents - 302046 92:92 - Other Miscellaneous Therapeutic Agents - 12:20.92 - Skeletal Muscle Relaxants, Miscellaneous - Adapalene 84:92 - Skin and Mucous Membrane Agents, Acalabrutinib 10:00 - Antineoplastic Agents - 317059 Adefovir Acamprosate 8:18.32 - Nucleosides and Nucleotides - 302036 28:92 - Central Nervous System Agents, Adenosine 24:04.04.24 - Class IV Antiarrhythmics - 304010 Acarbose Adenovirus Vaccine Live Oral 68:20.02 - alpha-Glucosidase Inhibitors - 396015 80:12 - Vaccines - 315016 Acebutolol Ado-Trastuzumab 24:24 - beta-Adrenergic Blocking Agents - 387003 10:00 - Antineoplastic Agents - 313041 12:16.08.08 - Selective -

Brincidofovir Is Highly Efficacious in Controlling Adenoviremia In
Brincidofovir is highly efficacious in controlling adenoviremia in pediatric recipients of hematopoietic cell transplant Prashant Hiwarkar,1 Persis Amrolia,2 Ponni Sivaprakasam,3 Su Han Lum,4 Hemalatha Doss,5 Ciara O’Rafferty,1 Toni Petterson,6 Katharine Patrick,7 Juliana Silva,2 Mary Slatter,5 Sarah Lawson,1 Kanchan Rao,2 Colin Steward,3 Adam Gassas,3 Paul Veys,2 Robert Wynn4 On behalf of the UK Paediatric BMT Group 1Department of Haematology and Bone Marrow Transplantation, Birmingham Children’s Hospital, Birmingham, UK; 2Department of Bone Marrow Transplantation, Great Ormond Street Hospital for Children, London; 3Department of Bone Marrow Transplantation, Royal Hospital for Children, Bristol, UK; 4Department of Bone Marrow Transplantation, Royal Manchester Children's Hospital, Manchester, UK; 5Department of Bone Marrow Transplantation, Great North Children's Hospital, Newcastle upon Tyne, UK; 6Department of Bone Marrow Transplantation, Royal Marsden Hospital, Sutton, UK; 7Department of Bone Marrow Transplantation, Sheffield Children's Hospital, Sheffield, UK Address for Correspondence: [email protected] Department of Haematology and Bone Marrow transplantation Birmingham Children’s Hospital, Steelhouse Lane, Birmingham, UK B4 6NH Tel. +44 (0)121 333 9999 Key points Brincidofovir has superior anti-adenoviral activity and safety profile compared to Cidofovir. Brincidofovir is highly efficacious in controlling adenoviraemia during lymphopenic phase of HCT. Summary Cidofovir is pre-emptively used for controlling adenoviremia and preventing disseminated viral disease in hematopoietic cell transplant (HCT) recipients but does not lead to resolution of viremia without T-cell immune-reconstitution. The lipid conjugated prodrug of Cidofovir, Brincidofovir, has improved oral bioavailability and achieves higher intracellular concentrations of active drug. -

Ebola Hemorrhagic Fever: Properties of the Pathogen and Development of Vaccines and Chemotherapeutic Agents O
ISSN 00268933, Molecular Biology, 2015, Vol. 49, No. 4, pp. 480–493. © Pleiades Publishing, Inc., 2015. Original Russian Text © O.I. Kiselev, A.V. Vasin, M.P. Shevyryova, E.G. Deeva, K.V. Sivak, V.V. Egorov, V.B. Tsvetkov, A.Yu. Egorov, E.A. RomanovskayaRomanko, L.A. Stepanova, A.B. Komissarov, L.M. Tsybalova, G.M. Ignatjev, 2015, published in Molekulyarnaya Biologiya, 2015, Vol. 49, No. 4, pp. 541–554. REVIEWS UDC 578.2,578.76 Ebola Hemorrhagic Fever: Properties of the Pathogen and Development of Vaccines and Chemotherapeutic Agents O. I. Kiseleva, A. V. Vasina, b, M. P. Shevyryovac, E. G. Deevaa, K. V. Sivaka, V. V. Egorova, V. B. Tsvetkova, d, A. Yu. Egorova, E. A. RomanovskayaRomankoa, L. A. Stepanovaa, A. B. Komissarova, L. M. Tsybalovaa, and G. M. Ignatjeva a Institute of Influenza, Ministry of Health of the Russian Federation, St. Petersburg, 197376 Russia; email: [email protected] b St. Petersburg State Polytechnic University, St. Petersburg, 195251 Russia c Ministry of Health of the Russian Federation, Moscow, 127994 Russia d Topchiev Institute of Petrochemical Synthesis, Moscow, 119991 Russia Received December 29, 2014; in final form, January 16, 2015 Abstract—Ebola hemorrhagic fever (EHF) epidemic currently ongoing in West Africa is not the first among numerous epidemics in the continent. Yet it seems to be the worst EHF epidemic outbreak caused by Ebola virus Zaire since 1976 as regards its extremely large scale and rapid spread in the population. Experiments to study the agent have continued for more than 20 years. The EHF virus has a relatively simple genome with seven genes and additional reading frame resulting from RNA editing. -

Keeping up with FDA Drug Approvals: 60 New Drugs in 60 Minutes Elizabeth A
Keeping Up with FDA Drug Approvals: 60 New Drugs in 60 Minutes Elizabeth A. Shlom, PharmD, BCPS Senior Vice President & Director Clinical Pharmacy Program | Acurity, Inc. Privileged and Confidential April 10, 2019 Privileged and Confidential Program Objectives By the end of the presentation, the pharmacist or pharmacy technician participant will be able to: ▪ Identify orphan drugs and first-in-class medications approved by the FDA in 2018. ▪ List five new drugs and their indications. ▪ Identify the place in therapy for three novel monoclonal antibodies. ▪ Discuss at least two new medications that address public health concerns. Dr. Shlom does not have any conflicts of interest in regard to this presentation. Both trade names and generic names will be discussed throughout the presentation Privileged and Confidential 2018 NDA Approvals (NMEs/BLAs) ▪ Lutathera (lutetium Lu 177 dotatate) ▪ Braftovi (encorafenib) ▪ Vizimpro (dacomitinib) ▪ Biktarvy (bictegravir, emtricitabine, ▪ TPOXX (tecovirimat) ▪ Libtayo (cemiplimab-rwic) tenofovir, ▪ Tibsovo (ivosidenib) ▪ Seysara (sarecycline) alafenamide) ▪ Krintafel (tafenoquine) ▪ Nuzyra (omadacycline) ▪ Symdeko (tezacaftor, ivacaftor) ▪ Orilissa (elagolix sodium) ▪ Revcovi (elapegademase-lvir) ▪ Erleada (apalutamide) ▪ Omegaven (fish oil triglycerides) ▪ Tegsedi (inotersen) ▪ Trogarzo (ibalizumab-uiyk) ▪ Mulpleta (lusutrombopag) ▪ Talzenna (talazoparib) ▪ Ilumya (tildrakizumab-asmn) ▪ Poteligeo (mogamulizumab-kpkc) ▪ Xofluza (baloxavir marboxil) ▪ Tavalisse (fostamatinib disodium) ▪ Onpattro (patisiran) -

Kinase Inhibitors and Nucleoside Analogues As Novel Therapies to Inhibit HIV-1 Or ZEBOV Replication
Kinase Inhibitors and Nucleoside Analogues as Novel Therapies to Inhibit HIV-1 or ZEBOV Replication by Stephen D. S. McCarthy A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Laboratory Medicine and Pathobiology University of Toronto © Copyright by Stephen D. S. McCarthy (2017) Kinase Inhibitors and Nucleoside Analogues as Novel Therapies to Inhibit HIV-1 or ZEBOV Replication Stephen D.S. McCarthy Doctor of Philosophy Graduate Department of Laboratory Medicine and Pathobiology University of Toronto 2017 Abstract Without a vaccine for Human Immunodeficiency Virus type 1 (HIV-1), or approved therapy for treating Zaire Ebolavirus (ZEBOV) infection, new means to treat either virus during acute infection are under intense investigation. Repurposing tyrosine kinase inhibitors of known specificity may not only inhibit HIV-1 replication, but also treat associated inflammation or neurocognitive disorders caused by chronic HIV-1 infection. Moreover, tyrosine kinase inhibitors may effectively treat other infections, including ZEBOV. In addition, established nucleoside/nucleotide analogues that effectively inhibit HIV-1 infection, could also be repurposed to inhibit ZEBOV replication. In this work the role of two host cell kinases, cellular protoncogene SRC (c-SRC) and Protein Tyrosine Kinase 2 Beta (PTK2B), were found to have key roles during early HIV-1 replication in primary activated CD4+ T-cells ex vivo. siRNA knockdown of either kinase increased intracellular reverse transcripts and decreased nuclear proviral integration, suggesting they act at the level of pre-integration complex (PIC) formation or PIC nuclear translocation. c-SRC siRNA knockdown consistently reduced p24 levels of IIIB(X4) and Ba-L(R5) infection, or luciferase ii activity of HXB2(X4) or JR-FL(R5) recombinant viruses, prompting further drug inhibition studies of this kinase. -

Situación Actual Del Tratamiento Farmacológico Frente a La Enfermedad Causada Por El Virus Ébola
Revisión Jordi Reina Situación actual del tratamiento farmacológico frente a la enfermedad causada por el virus Ébola Unidad de Virología. Servicio de Microbiología. Hospital Universitario Son Espases. Palma de Mallorca RESUMEN Current status of drug treatment against the disease caused by the Ebola virus La reciente epidemia de enfermedad causada por el virus Ébola ha puesto en evidencia la necesidad de desarrollar y dis- ABSTRACT poner de fármacos específicos para hacer frente a esta entidad. De acuerdo con los datos virológicos se han diseñado algunos The recent epidemic of disease caused by the Ebola virus fármacos nuevos y se ha comprobado que otros podrían tener has highlighted the need to develop specific drugs and have to eficacia frente a este virus. deal with this entity. According to virological analysis they have Las principales líneas terapéuticas se basan en la inmu- been designed to give you some new drugs and are proven to noterapia (suero de pacientes convalescientes y anticuerpos others might be effective against this virus. monoclonales específicos), en fármacos antivirales (favipiora- The main lines of therapy are based on immunotherapy vir, BCX4430, Brincidofovir), RNAs de interferencia (TKM-Ébola) (convalescent serum of patients and specific monoclonal an- y oligonucleótidos sin sentido (morfolino fosforodiamidato) y tibodies), antiviral drugs (favipiravir, BCX4430, brincidofovir), otros fármacos no antivirales (clomifeno, NSC62914, FGI-103, interfering RNAs (TKM-Ebola) and antisense oligonucleotides amilorida y la ouabaina). (morpholino phosphorodiamidate) and other drugs no antiviral Los estudios existentes son escasos y principalmente en (clomiphene NSC62914, FGI-103, amiloride and ouabain). modelos animales y los ensayos clínicos han quedado la ma- Existing studies are scarce and mainly in animal models yoría inconclusos por la disminución drástica del número de and clinical trials have been inconclusive most by the drastic nuevos casos. -

Integrated Computational Approach for Virtual Hit Identification Against
International Journal of Molecular Sciences Article Integrated Computational Approach for Virtual Hit Identification against Ebola Viral Proteins VP35 and VP40 Muhammad Usman Mirza 1,2,*,† and Nazia Ikram 2 1 Centre for Research in Molecular Medicine (CRiMM), The University of Lahore, Defense Road, Lahore 54000, Pakistan 2 Institute of Molecular Biology and Biotechnology, The University of Lahore, Lahore 54000, Pakistan; [email protected] * Correspondence: [email protected] or [email protected]; Tel.: +92-333-839-6037 † Current address: Department of Pharmaceutical and Pharmacological Sciences, Rega Institute for Medical Research, Medicinal Chemistry, University of Leuven, Leuven B-3000, Belgium. Academic Editors: Christo Z. Christov and Tatyana Karabencheva-Christova Received: 28 July 2016; Accepted: 22 September 2016; Published: 26 October 2016 Abstract: The Ebola virus (EBOV) has been recognised for nearly 40 years, with the most recent EBOV outbreak being in West Africa, where it created a humanitarian crisis. Mortalities reported up to 30 March 2016 totalled 11,307. However, up until now, EBOV drugs have been far from achieving regulatory (FDA) approval. It is therefore essential to identify parent compounds that have the potential to be developed into effective drugs. Studies on Ebola viral proteins have shown that some can elicit an immunological response in mice, and these are now considered essential components of a vaccine designed to protect against Ebola haemorrhagic fever. The current study focuses on chemoinformatic approaches to identify virtual hits against Ebola viral proteins (VP35 and VP40), including protein binding site prediction, drug-likeness, pharmacokinetic and pharmacodynamic properties, metabolic site prediction, and molecular docking. Retrospective validation was performed using a database of non-active compounds, and early enrichment of EBOV actives at different false positive rates was calculated. -

A Library of Nucleotide Analogues Terminate RNA Synthesis Catalyzed by Polymerases of Coronaviruses Causing SARS and COVID-19
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.23.058776; this version posted April 25, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A Library of Nucleotide Analogues Terminate RNA Synthesis Catalyzed by Polymerases of Coronaviruses Causing SARS and COVID-19 Steffen Jockusch1,2,#, Chuanjuan Tao1,3,#, Xiaoxu Li1,3,#, Thomas K. Anderson5,6, Minchen Chien1,3, Shiv Kumar1,3, James J. Russo1,3, Robert N. Kirchdoerfer5,6,*, Jingyue Ju1,3,4,* 1Center for Genome Technology and Biomolecular Engineering, Columbia University, New York, NY 10027; Departments of 2 Chemistry, 3Chemical Engineering, and 4Pharmacology, Columbia University, New York, NY 10027; 5Departments of Biochemistry and 6Institute of Molecular Virology, University of Wisconsin-Madison, Madison, WI 53706 #SJ, CT and XL contributed equally to this work. *To whom correspondence should be addressed: [email protected] (JJ); [email protected] (RNK) Abstract SARS-CoV-2, a member of the coronavirus family, is responsible for the current COVID-19 worldwide pandemic. We previously demonstrated that five nucleotide analogues inhibit the SARS-CoV-2 RNA- dependent RNA polymerase (RdRp), including the active triphosphate forms of Sofosbuvir, Alovudine, Zidovudine, Tenofovir alafenamide and Emtricitabine. We report here the evaluation of a library of additional nucleoside triphosphate analogues with a variety of structural and chemical features as inhibitors of the RdRps of SARS-CoV and SARS-CoV-2. These features include modifications on the sugar (2’ or 3’ modifications, carbocyclic, acyclic, or dideoxynucleotides) or on the base. -

Student Clinical Digest
2014-2015 STUDENT CLINICAL DIGEST Presented by the Student College of Clinical Pharmacy— the official UGA chapter of American College of Clinical Pharmacy Volume 1, Issue 4 Clinical Pharmacist Focus ences and humble advice to our tered. I wanted to be a generalist students. and didn’t want to specialize out of fear of being too narrowly Why did you decide to pursue a focused and lacking variety. I residency? liked being able to dip my feet in other fields, and this has really I decided to pursue a residency helped me in the positions I’ve because of the opportunities held. available to me as a 4th year stu- dent on rotation at Emory Uni- What made you switch from versity Medical Center. During hospital to academia? this APPE, it was the 1st time I Michael Neville, Pharm. D., saw pharmacist working with the Someone told me many years ago BCPS, FASHP surgery team, internal medicine, they thought I would be a really intensive care unit, and transplant. great teacher and other people INSIDE THIS By Khushbu Tejani, Pharm D. Candi- What really impressed me was pointed it out a few more times ISSUE: date seeing the pharmacists being during my career. Both my par- proactive and contributing to ents come from an educational In this issue, we meet Dr. Michael patient care in a way that I never background and wanted me to Neville, a clinical pharmacist as saw before. While there, they got follow. I initially did not want to Clinical 1 well as former Clinical Associate to know me and asked me to pursue academia but it runs in my Pharmacist Focus Professor at the College of Phar- consider applying for their resi- blood.