OR1S2 (NM 001004459) Human Untagged Clone – SC300694
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -
Supplementary Information
Supplementary Information Table S1. Regions of CNV data excluded from CNV analysis due to poor density of probe coverage. Region Centromere Telomere Chr. p q p q 1 p11.1, p11.2, q11, q12, q21.1 p36.33, p36.32, p36.31, q44 p12, p13.1 p36.23, p36.22, p36.21 2 p11.1, p11.2 q11.1 p25.3 q37.3 3 p11.1 q11.1, q11.2 none excluded q29 4 p11 q11 p16.1, p16.2, p16.3 q35.2, q35.1 5 p12 q11.1 p15.33 q35.3, q35.2, q35.1 6 p11.1 q11.1 p25.3, p35.2 q27 7 p11.1 q11.1, q11.21 p22.3 q36.3 8 p11.1 q11.1 p23.3, p23.2, p23.1 q24.3 9 p11.1, p11.2 q11, q12, q13 p24.3 q34.3, q34.2, q34.13, q34.12, q34.11 10 p11.1 q11.1, q11.21, q11.22 none excluded q26.3 11 p11.11, p11.12 q11 p15.5 q25 12 p11.1, p11.21 q11 p13.33, p13.32 q24.33, q24.32, q24.31 13 no probes q11 no probes q34 14 no probes q11.1, q11.2 no probes q32.33 15 no probes q11.1, q11.2, q12, no probes q26.3 q13.1, q13.2, q13.3 16 p11.1, p11.2 q11.1, q11.2 p13.3 q24.3, q24.2, q24.1 17 none excluded none excluded p13.3 q25.3 18 p11.1, p11.21 q11.1 none excluded q23 19 p11 q11 p13.3 q13.43 20 p11.1, p11.21 q11.1, q11.21 none excluded q13.33 21 no probes q11.1, q11.2 no probes q22.3 22 no probes q11.1, q11.21, q11.22 no probes q13.33, q13.32 Table S2. -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -

Cellular Features Predicting Susceptibility to Ferroptosis: Insights from Cancer Cell-Line Profiling
Cellular features predicting susceptibility to ferroptosis: insights from cancer cell-line profiling Vasanthi Sridhar Viswanathan Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2015 © 2015 Vasanthi Sridhar Viswanathan All rights reserved ABSTRACT Cellular features predicting susceptibility to ferroptosis: insights from cancer cell-line profiling Vasanthi Sridhar Viswanathan Ferroptosis is a novel non-apoptotic, oxidative form of regulated cell death that can be triggered by diverse small-molecule ferroptosis inducers (FINs) and genetic perturbations. Current lack of insights into the cellular contexts governing sensitivity to ferroptosis has hindered both translation of FINs as anti-cancer agents for specific indications and the discovery of physiological contexts where ferroptosis may function as a form of programmed cell death. This dissertation describes the identification of cellular features predicting susceptibility to ferroptosis from data generated through a large-scale profiling experiment that screened four FINs against a panel of 860 omically-characterized cancer cell lines (Cancer Therapeutics Response Portal Version 2; CTRPv2 at http://www.broadinstitute.org/ctrp/). Using correlative approaches incorporating transcriptomic, metabolomic, proteomic, and gene-dependency feature types, I uncover both pan-lineage and lineage-specific features mediating cell-line response to FINs. The first key finding from these analyses implicates high expression of sulfur and selenium metabolic pathways in conferring resistance to FINs across lineages. In contrast, the transsulfuration pathway, which enables de novo cysteine synthesis, appears to plays a role in ferroptosis resistance in a subset of lineages. The second key finding from these studies identifies cancer cells in a high mesenchymal state as being uniquely primed to undergo ferroptosis. -

Analysis of RHOA Mutations and Their Significance in the Proliferation and Transcriptome of Digestive Tract Cancer Cells
ONCOLOGY LETTERS 22: 735, 2021 Analysis of RHOA mutations and their significance in the proliferation and transcriptome of digestive tract cancer cells NAOKI IKARI1,2, AKIKO SERIZAWA1, ETSUKO TANJI2, MASAKAZU YAMAMOTO1 and TORU FURUKAWA1‑3 1Department of Surgery, Institute of Gastroenterology; 2Institute for Integrated Medical Sciences, Tokyo Women's Medical University, Shinjuku‑ku, Tokyo 162‑8666; 3Department of Investigative Pathology, Tohoku University Graduate School of Medicine, Aoba‑ku, Sendai 980‑8575, Japan Received April 23, 2021; Accepted July 14, 2021 DOI: 10.3892/ol.2021.12996 Abstract. The ras homolog family member A (RHOA) gene showed that the genes associated with small molecule metabolic encodes a member of the Rho family of small GTPases and is process, oxidation‑reduction processes, protein kinase activity, known to function in reorganization of the actin cytoskeleton, transport, and cell junction were commonly downregulated in which is associated with regulation of cell shape, attachment cells whose proliferation was attenuated by the knockdown of and motility. RHOA has been found to be recurrently mutated RHOA. These results suggested that certain RHOA mutations in gastrointestinal cancer; however, the functional signifi‑ may result in upregulation of lnc‑DERA‑1 and genes associated cance of the mutated RHOA protein in digestive tract cancers with cellular metabolism and proliferation in digestive tract remains to be uncovered. The aim of the present study was cancers. to understand the role of mutant RHOA in the proliferation and transcriptome of digestive tract cancer cells. Mutations of Introduction RHOA in one esophageal cancer cell line, OE19, eight gastric cancer cell lines, namely, AGS, GCIY, HGC‑27, KATO III, The ras homolog family member A (RHOA) gene encodes a MKN1, MKN45, SNU16 and SNU719, as well as two colon member of the Rho family of small GTPases and is known cancer cell lines, CCK‑81 and SW948, were determined using to function in reorganization of the actin cytoskeleton, which Sanger sequencing. -

Quadruple Contextfree Lsystem M Athe M Atical Tools As Origin Of
1 Quadruple context-free L-System mathematical tools as origin of biological evolution Arunava Goswa mi $, Pabitra Pal Choudhury #, Rajneesh Singh$, #, Sk. Sarif Hassan$, # $Biological Sciences Division and #Applied Statistics Unit, Indian Statistical Institute, 203, B. T. Road, Kolkata 700108, India It is well known that A, T, G, C annealed together early in evolution and the long stretch of DNA was found which ultimately resulted into chromosomes of different organisms. But it is unclear till date how exons, introns, conserved protein domains was formed. Using the DNA sequences of the largest known gene-family present in human genome, i.e., olfactory receptors and simplest possible quadruple context-free L-Systems, we show that conserved protein domains and intergenic regions which lies at the heart of the biological evolution started with a sixteen base-pairs stretch of DNA. 1. Use of quadruple context-free L-Systems on olfactory receptors (OR) ORs constitute the largest known multigene family involved in sense of smell of different organisms. There are ~388 functional and ~414 pseudo OR genes are present in human genome [1]. Researchers have found that except two chromosomes, ORs are unevenly distributed in 21 chromosomes. OR1 family contains 28 full lengths and 5 pseudo ORs as per HORDE database (https://senselab.med.yale.edu/ORDB/files/humanorseqanal.html). Fig. 1 shows the ClustalW (http://align.genome.jp/) data of extreme 5'-end stretch of 29 full length ORs. Fig. 1 Nature Precedings : doi:10.1038/npre.2010.4851.1 Posted 2 Sep -
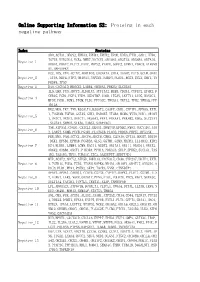
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2 -

A Gain-Of-Function Mutation in the GRIK2 Gene Causes Neurodevelopmental Deficits
A gain-of-function mutation in the GRIK2 gene causes neurodevelopmental deficits Yomayra F. Guzmán, ABSTRACT PhD Objective: To identify inherited or de novo mutations associated with a suite of neurodevelopmen- Keri Ramsey, RN, tal abnormalities in a 10-year-old patient displaying ataxia, motor and speech delay, and intellec- * CCRN tual disability. Jacob R. Stolz, BA Methods: We performed whole-exome sequencing of the proband and her parents. A pathogenic David W. Craig, PhD* gene variant was identified as damaging based on sequence conservation, gene function, and Mathew J. Huentelman, association with disorders having similar phenotypic profiles. Functional characterization of the PhD* mutated protein was performed in vitro using a heterologous expression system. Vinodh Narayanan, MD* GRIK2 Geoffrey T. Swanson, Results: A single de novo point mutation in the gene was identified as causative for the PhD neurologic symptoms of the proband. The mutation is predicted to change a codon for alanine to On behalf of the C4RCD that of a threonine at position 657 (A657T) in the GluK2 kainate receptor (KAR) subunit, a mem- Research Group ber of the ionotropic glutamate receptor gene family. Whole-cell voltage-clamp recordings re- vealed that KARs incorporating the GluK2(A657T) subunits show profoundly altered channel gating and are constitutively active in nominally glutamate-free extracellular media. Correspondence to Conclusions: In this study, we associate a de novo gain-of-function mutation in the GRIK2 gene Dr. Swanson: [email protected] with deficits in motor and higher order cognitive function. These results suggest that disruption of physiologic KAR function precludes appropriate development of the nervous system. -

Identifying Drug Sensitivity Subnetworks with NETPHIX
bioRxiv preprint doi: https://doi.org/10.1101/543876. this version posted February 8, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. It is made available under a CC-BY-NC-ND 4.0 International license. Identifying Drug Sensitivity Subnetworks with NETPHLIX Yoo-Ah Kim *† Rebecca Sarto Basso *†‡ Damian Wojtowicz † Dorit S. Hochbaum ‡ Fabio Vandin §¶ Teresa M. Prztycka †¶ Abstract Phenotypic heterogeneity in cancer is often caused by different patterns of genetic alterations. Under- standing such phenotype-genotype relationships is fundamental for the advance of personalized medicine. One of the important challenges in the area is to predict drug response on a personalized level. The pathway- centric view of cancer significantly advanced the understanding of genotype-phenotype relationships. How- ever, most of network identification methods in cancer focus on identifying subnetworks that include gen- eral cancer drivers or are associated with discrete features such as cancer subtypes, hence cannot be applied directly for the analysis of continuous features like drug response. On the other hand, existing genome wide association approaches do not fully utilize the complex proprieties of cancer mutational landscape. To ad- dress these challenges, we propose a computational method, named NETPHLIX (NETwork-to-PHenotpe mapping LeveragIng eXlusivity), which aims to identify mutated subnetworks that are associated with drug response (or any continuous cancer phenotype). Utilizing properties such as mutual exclusivity and interac- tions among genes, we formulate the problem as an integer linear program and solve it optimally to obtain a set of genes satisfying the constraints. NETPHLIX identified gene modules significantly associated with many drugs, including interesting response modules to MEK1/2 inhibitors in both directions (increased and decreased sensitivity to the drug) that the previous method, which does not utilize network informa- tion, failed to identify. -

Supplementary Methods
doi: 10.1038/nature06162 SUPPLEMENTARY INFORMATION Supplementary Methods Cloning of human odorant receptors 423 human odorant receptors were cloned with sequence information from The Olfactory Receptor Database (http://senselab.med.yale.edu/senselab/ORDB/default.asp). Of these, 335 were predicted to encode functional receptors, 45 were predicted to encode pseudogenes, 29 were putative variant pairs of the same genes, and 14 were duplicates. We adopted the nomenclature proposed by Doron Lancet 1. OR7D4 and the six intact odorant receptor genes in the OR7D4 gene cluster (OR1M1, OR7G2, OR7G1, OR7G3, OR7D2, and OR7E24) were used for functional analyses. SNPs in these odorant receptors were identified from the NCBI dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP) or through genotyping. OR7D4 single nucleotide variants were generated by cloning the reference sequence from a subject or by inducing polymorphic SNPs by site-directed mutagenesis using overlap extension PCR. Single nucleotide and frameshift variants for the six intact odorant receptors in the same gene cluster as OR7D4 were generated by cloning the respective genes from the genomic DNA of each subject. The chimpanzee OR7D4 orthologue was amplified from chimpanzee genomic DNA (Coriell Cell Repositories). Odorant receptors that contain the first 20 amino acids of human rhodopsin tag 2 in pCI (Promega) were expressed in the Hana3A cell line along with a short form of mRTP1 called RTP1S, (M37 to the C-terminal end), which enhances functional expression of the odorant receptors 3. For experiments with untagged odorant receptors, OR7D4 RT and S84N variants without the Rho tag were cloned into pCI. -

Research/0018.1
http://genomebiology.com/2001/2/6/research/0018.1 Research The human olfactory receptor repertoire comment Sergey Zozulya, ernando Echeverri and Trieu Nguyen Address: Senomyx Inc., 11099 North Torrey Pines Road, La Jolla, CA 92037, USA. Correspondence: Sergey Zozulya. E-mail: [email protected] reviews Published: 1 June 2001 Received: 8 March 2001 Revised: 12 April 2001 Genome Biology 2001, 2(6):research0018.1–0018.12 Accepted: 18 April 2001 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2001/2/6/research/0018 © 2001 Zozulya et al., licensee BioMed Central Ltd (Print ISSN 1465-6906; Online ISSN 1465-6914) reports Abstract Background: The mammalian olfactory apparatus is able to recognize and distinguish thousands of structurally diverse volatile chemicals. This chemosensory function is mediated by a very large family of seven-transmembrane olfactory (odorant) receptors encoded by approximately 1,000 genes, the majority of which are believed to be pseudogenes in humans. deposited research Results: The strategy of our sequence database mining for full-length, functional candidate odorant receptor genes was based on the high overall sequence similarity and presence of a number of conserved sequence motifs in all known mammalian odorant receptors as well as the absence of introns in their coding sequences. We report here the identification and physical cloning of 347 putative human full-length odorant receptor genes. Comparative sequence analysis of the predicted gene products allowed us to identify and define a number of consensus sequence motifs and structural features of this vast family of receptors. -

Association of Somatic Mutations of ADAMTS Genes with Improved Chemotherapy Sensitivity and Survival in High-Grade Serous Ovarian Carcinoma
Supplementary Online Content Liu Y, Yasukawa M, Chen K, et al. Association of somatic mutations of ADAMTS genes with improved chemotherapy sensitivity and survival in high-grade serous ovarian carcinoma. JAMA Oncol. Published online June 11, 2015. doi:10.1001/jamaoncol.2015.1432 eMethods eFigure 1. Analysis flow chart for identification and validation of ADAMTS mutations eFigure 2. Responder-related genes identified in the TCGA discovery cohort eFigure 3. Distribution of protein alterations encoded in the ADAMTS genes in the TCGA discovery cohort eFigure 4. Association of BRCA mutations with survival and chemotherapy response in the TCGA discovery cohort eFigure 5. Association of residual tumor size with survival and chemotherapy response in the TCGA discovery cohort. eFigure 6. Association of ADAMTS mutations with genetic instability in the TCGA discovery cohort. eFigure 7. Comparison of association of ADAMTS and BRCA1/2 mutations with mutation frequency in the TCGA discovery cohort eFigure 8. Association of ADAMTS mutations with mutation spectra in the TCGA discovery cohort eFigure 9. Correlation of mutation frequency with C>T or A>T fractions in the TCGA discovery cohort eFigure 10. Distribution of the number of patients harboring gene mutations among the 105 random gene selections eFigure 11. Statistical significance (P value) of outcome association among the 105 random gene selections eFigure 12. Distribution of OvCa patients from different tissue source sites in the TCGA discovery cohort and validation cohort eFigure 13. ADAMTS and BRCA1/2 mutations in the TCGA validation cohort © 2015 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/27/2021 eFigure 14.