Urea Amidolyase of Candida Utilis Characterization Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Food Microbiology Influence of Nitrogen Status in Wine Alcoholic Fermentation
Food Microbiology 83 (2019) 71–85 Contents lists available at ScienceDirect Food Microbiology journal homepage: www.elsevier.com/locate/fm Influence of nitrogen status in wine alcoholic fermentation T ∗ Antoine Goberta, , Raphaëlle Tourdot-Maréchala, Céline Sparrowb, Christophe Morgeb, Hervé Alexandrea a UMR Procédés Alimentaires et Microbiologiques, Université de Bourgogne Franche-Comté/ AgroSup Dijon - Equipe VAlMiS (Vin, Aliment, Microbiologie, Stress), Institut Universitaire de la Vigne et du Vin Jules Guyot, Université de Bourgogne, Dijon, France b SAS Sofralab, 79, Av. A.A. Thévenet, BP 1031, Magenta, France ARTICLE INFO ABSTRACT Keywords: Nitrogen is an essential nutrient for yeast during alcoholic fermentation. Nitrogen is involved in the biosynthesis Nitrogen of protein, amino acids, nucleotides, and other metabolites, including volatile compounds. However, recent Amino acids studies have called several mechanisms that regulate its role in biosynthesis into question. An initial focus on S. Ammonium cerevisiae has highlighted that the concept of “preferred” versus “non-preferred” nitrogen sources is extremely Alcoholic fermentation variable and strain-dependent. Then, the direct involvement of amino acids consumed in the formation of Yeasts proteins and volatile compounds has recently been reevaluated. Indeed, studies have highlighted the key role of Wine Volatile compounds lipids in nitrogen regulation in S. cerevisiae and their involvement in the mechanism of cell death. New wine- making strategies using non-Saccharomyces yeast strains in co- or sequential fermentation improve nitrogen management. Indeed, recent studies show that non-Saccharomyces yeasts have significant and specific needs for nitrogen. Moreover, sluggish fermentation can occur when they are associated with S. cerevisiae, necessitating nitrogen addition. In this context, we will present the consequences of nitrogen addition, discussing the sources, time of addition, transcriptome changes, and effect on volatile compound composition. -

Lifestyle Adaptations of Rhizobium from Rhizosphere to Symbiosis
Lifestyle adaptations of Rhizobium from rhizosphere to symbiosis Rachel M. Wheatleya,1, Brandon L. Forda,1,LiLib,1, Samuel T. N. Aroneya, Hayley E. Knightsa, Raphael Ledermanna, Alison K. Easta, Vinoy K. Ramachandrana,2, and Philip S. Poolea,2 aDepartment of Plant Sciences, University of Oxford, OX1 3RB Oxford, United Kingdom; and bChinese Academy of Sciences Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, 430074 Wuhan, People’s Republic of China Edited by Éva Kondorosi, Hungarian Academy of Sciences, Biological Research Centre, Szeged, Hungary, and approved August 4, 2020 (received for review May 7, 2020) By analyzing successive lifestyle stages of a model Rhizobium– nodule cells and undergo terminal differentiation into N2-fixing legume symbiosis using mariner-based transposon insertion se- bacteroids (10). Nodules provide a protective microaerobic envi- quencing (INSeq), we have defined the genes required for rhizo- ronment to maintain oxygen-labile nitrogenase (6). In exchange + sphere growth, root colonization, bacterial infection, N2-fixing for NH4 and alanine, the legume host provides carbon sources to bacteroids, and release from legume (pea) nodules. While only 27 fuel this process, primarily as dicarboxylic acids (13, 14). genes are annotated as nif and fix in Rhizobium leguminosarum,we However, nodule infection is only one stage of the lifestyle of show 603 genetic regions (593 genes, 5 transfer RNAs, and 5 RNA rhizobia, and they spend much of their time surviving in the rhi- features) are required for the competitive ability to nodulate pea and zosphere, the zone of soil immediately surrounding roots (15). -

Site-Directed Mutagenesis Studies of E. Coli Biotin Carboxylase Valerie Melissa Sloane Louisiana State University and Agricultural and Mechanical College
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 2004 Site-directed mutagenesis studies of E. coli biotin carboxylase Valerie Melissa Sloane Louisiana State University and Agricultural and Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Recommended Citation Sloane, Valerie Melissa, "Site-directed mutagenesis studies of E. coli biotin carboxylase" (2004). LSU Doctoral Dissertations. 1561. https://digitalcommons.lsu.edu/gradschool_dissertations/1561 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. SITE-DIRECTED MUTAGENESIS STUDIES OF E. COLI BIOTIN CARBOXYLASE A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Biological Sciences by Valerie M. Sloane B.S., University of Louisiana at Lafayette, 1995 May 2004 ACKNOWLEDGEMENTS I would like to thank Dr. Grover Waldrop, my major professor, for his invaluable guidance and helpful insights throughout this project, as well as for obtaining grants for the research. I am also indebted to Professor Simon Chang, Associate Professor Patrick DiMario, Associate Professor Jacqueline Stephens, Assistant Professor Anne Grove, and Assistant Professor Britt Thomas for serving on my committee and for a thoughtful reading of each chapter. Dr. Carol Blanchard and Tee Bordelon especially deserve my thanks for their careful tutelage in molecular biology and protein purification techniques. -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0 -

Genome-Wide Investigation of Cellular Functions for Trna Nucleus
Genome-wide Investigation of Cellular Functions for tRNA Nucleus- Cytoplasm Trafficking in the Yeast Saccharomyces cerevisiae DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Hui-Yi Chu Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2012 Dissertation Committee: Anita K. Hopper, Advisor Stephen Osmani Kurt Fredrick Jane Jackman Copyright by Hui-Yi Chu 2012 Abstract In eukaryotic cells tRNAs are transcribed in the nucleus and exported to the cytoplasm for their essential role in protein synthesis. This export event was thought to be unidirectional. Surprisingly, several lines of evidence showed that mature cytoplasmic tRNAs shuttle between nucleus and cytoplasm and their distribution is nutrient-dependent. This newly discovered tRNA retrograde process is conserved from yeast to vertebrates. Although how exactly the tRNA nuclear-cytoplasmic trafficking is regulated is still under investigation, previous studies identified several transporters involved in tRNA subcellular dynamics. At least three members of the β-importin family function in tRNA nuclear-cytoplasmic intracellular movement: (1) Los1 functions in both the tRNA primary export and re-export processes; (2) Mtr10, directly or indirectly, is responsible for the constitutive retrograde import of cytoplasmic tRNA to the nucleus; (3) Msn5 functions solely in the re-export process. In this thesis I focus on the physiological role(s) of the tRNA nuclear retrograde pathway. One possibility is that nuclear accumulation of cytoplasmic tRNA serves to modulate translation of particular transcripts. To test this hypothesis, I compared expression profiles from non-translating mRNAs and polyribosome-bound translating mRNAs collected from msn5Δ and mtr10Δ mutants and wild-type cells, in fed or acute amino acid starvation conditions. -

The Ubiquitin Ligase Ariadne-1 Regulates NSF for Neurotransmitter Release
bioRxiv preprint doi: https://doi.org/10.1101/2020.01.23.916619; this version posted January 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The ubiquitin ligase Ariadne-1 regulates NSF for neurotransmitter release Juanma Ramírez1, Miguel Morales2#, Nerea Osinalde3, Imanol Martínez-Padrón2 Ugo Mayor1,4,* and Alberto Ferrús2,* From the 1Department of Biochemistry and Molecular Biology, Faculty of Science and Technology, UPV/EHU, Leioa 48940, Bizkaia, Spain; 2Cajal Institute, CSIC, Madrid 28002, Spain; 3Department of Biochemistry and Molecular Biology, Faculty of Pharmacy, UPV/EHU, Vitoria-Gasteiz 01006, Araba, Spain; 4Ikerbasque, Basque Foundation for Science, Bilbao 48013, Bizkaia, Spain Running title: Ubiquitinated synaptic NSF #Molecular Cognition Laboratory, Biophysics Institute UPV/EHU-CSIC, Barrio Sarriena, Leioa 48940, Bizkaia, Spain *To whom the correspondence should be addressed: Alberto Ferrús: Cajal Institute, CSIC, Madrid 28002, Spain; [email protected]; Ugo Mayor: Department of Biochemistry and Molecular Biology, Faculty of Science and Technology, UPV/EHU, Leioa 48940, Bizkaia, Spain; [email protected]. Keywords: Ariadne-1, Drosophila, E3 ubiquitin ligase, neurotransmitter release, NSF, synapse, ubiquitination ABSTRACT mechanism to regulate NSF activity in the synapse Ariadne-1 (Ari-1) is an essential E3 ubiquitin- through Ari-1-dependent ubiquitination. ligase whose neuronal substrates are yet to be identified. We have used an in vivo ubiquitin biotinylation strategy coupled to quantitative Neurotransmitter release is mediated by a set proteomics to identify putative Ari-1 substrates in of protein-protein interactions that include the N- Drosophila heads. -

WO 2014/202616 A2 24 December 2014 (24.12.2014) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2014/202616 A2 24 December 2014 (24.12.2014) P O P C T (51) International Patent Classification: 13 172714 1 I June 2013 (19.06.2013) EP C07K 14/37 (2006.01) 13 172724 0 I June 2013 (19.06.2013) EP 13 172685 3 I June 2013 (19.06.2013) EP (21) International Application Number: 13 172686 1 I S June 2013 (19.06.2013) EP PCT/EP2014/062737 13 172683 8 I S June 2013 (19.06.2013) EP (22) International Filing Date: 13 172672 1 I S June 2013 (19.06.2013) EP 17 June 2014 (17.06.2014) 13 172673 9 I S June 2013 (19.06.2013) EP 13 172675 4 I S June 2013 (19.06.2013) EP (25) Filing Language: English 13 172677 0 I S June 2013 (19.06.2013) EP (26) Publication Lan ua e: English 13 172681 2 I S June 2013 (19.06.2013) EP 13 172837 0 I S June 2013 (19.06.2013) EP (30) Priority Data: 13 17261 1 9 I S June 2013 (19.06.2013) EP 13 172700.0 19 June 2013 (19 .06.2013) EP 13 172784 4 I S June 2013 (19.06.2013) EP 13 172812.3 19 June 2013 (19 .06.2013) EP 13 172821 4 I S June 2013 (19.06.2013) EP 13 172758.8 19 June 2013 (19 .06.2013) EP 13 172615 0 I S June 2013 (19.06.2013) EP 13 172757.0 19 June 2013 (19 .06.2013) EP 13 172624 2 I S June 2013 (19.06.2013) EP 13 172842.0 19 June 2013 (19 .06.2013) EP 13 172680 4 I S June 2013 (19.06.2013) EP 13 172756.2 19 June 2013 (19 .06.2013) EP 13 172623 4 I S June 2013 (19.06.2013) EP 13 172759.6 -

Non-Saccharomyces Yeasts and Organic Wines Fermentation: Implications on Human Health
fermentation Review Non-Saccharomyces Yeasts and Organic Wines Fermentation: Implications on Human Health Alice Vilela CQ-VR, Chemistry Research Centre, School of Life Sciences and Environment, Dep. of Biology and Environment, University of Trás-os-Montes and Alto Douro (UTAD), 5000-801 Vila Real, Portugal; [email protected] Received: 12 May 2020; Accepted: 21 May 2020; Published: 25 May 2020 Abstract: A relevant trend in winemaking is to reduce the use of chemical compounds in both the vineyard and winery. In organic productions, synthetic chemical fertilizers, pesticides, and genetically modified organisms must be avoided, aiming to achieve the production of a “safer wine”. Safety represents a big threat all over the world, being one of the most important goals to be achieved in both Western society and developing countries. An occurrence in wine safety results in the recovery of a broad variety of harmful compounds for human health such as amines, carbamate, and mycotoxins. The perceived increase in sensory complexity and superiority of successful uninoculated wine fermentations, as well as a thrust from consumers looking for a more “natural” or “organic” wine, produced with fewer additives, and perceived health attributes has led to more investigations into the use of non-Saccharomyces yeasts in winemaking, namely in organic wines. However, the use of copper and sulfur-based molecules as an alternative to chemical pesticides, in organic vineyards, seems to affect the composition of grape microbiota; high copper residues can be present in grape must and wine. This review aims to provide an overview of organic wine safety, when using indigenous and/or non-Saccharomyces yeasts to perform fermentation, with a special focus on some metabolites of microbial origin, namely, ochratoxin A (OTA) and other mycotoxins, biogenic amines (BAs), and ethyl carbamate (EC). -
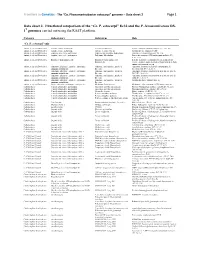
Ca. P. Ectocarpi” Ec32 and the P
Frontiers in Genetics - The “Ca. Phaeomarinobacter ectocarpi” genome – Data sheet 2 Page 1 Data sheet 2. Functional comparison of the “Ca. P. ectocarpi” Ec32 and the P. lavamentivorans DS- 1T genomes carried out using the RAST platform. Category Subcategory Subsystem Role “Ca. P. ectocarpi” only Amino Acids and Derivatives Alanine, serine, and glycine Alanine biosynthesis Valine--pyruvate aminotransferase (EC 2.6.1.66) Amino Acids and Derivatives Alanine, serine, and glycine Glycine cleavage system Sodium/glycine symporter GlyP Amino Acids and Derivatives Arginine; urea cycle, polyamines Arginine and Ornithine Degradation Ornithine cyclodeaminase (EC 4.3.1.12) Amino Acids and Derivatives Arginine; urea cycle, polyamines Polyamine Metabolism Putrescine transport ATP-binding protein PotA (TC 3.A.1.11.1) Amino Acids and Derivatives Branched-chain amino acids Branched-Chain Amino Acid Leucine-responsive regulatory protein, regulator for Biosynthesis leucine (or lrp) regulon and high-affinity branched-chain amino acid transport system Amino Acids and Derivatives Glutamine, glutamate, aspartate, asparagine; Glutamate and Aspartate uptake in Glutamate Aspartate periplasmic binding protein ammonia assimilation Bacteria precursor GltI (TC 3.A.1.3.4) Amino Acids and Derivatives Glutamine, glutamate, aspartate, asparagine; Glutamate and Aspartate uptake in Glutamate Aspartate transport system permease protein ammonia assimilation Bacteria GltJ (TC 3.A.1.3.4) Amino Acids and Derivatives Glutamine, glutamate, aspartate, asparagine; Glutamate and Aspartate -

Molecular Diagnostic Testing Requisition
BAYLOR GENETICS PHONE CONNECT 2450 HOLCOMBE BLVD. 1.800.411.4363 GRAND BLVD. RECEIVING DOCK FAX HOUSTON, TX 77021-2024 1.800.434.9850 MOLECULAR DIAGNOSTIC TESTING REQUISITION PATIENT INFORMATION (COMPLETE ONE FORM FOR EACH PERSON TESTED) / / Patient Last Name Patient First Name MI Date of Birth (MM / DD / YYYY) Address City State Zip Phone Patient discharged from Biological Sex: the hospital/facility: Female Male Unknown Accession # Hospital / Medical Record # Yes No Gender identity (if diff erent from above): REPORTING RECIPIENTS Ordering Physician Institution Name Email (Required for International Clients) Phone Fax ADDITIONAL RECIPIENTS Name Email Fax Name Email Fax PAYMENT (FILL OUT ONE OF THE OPTIONS BELOW) SELF PAYMENT Pay With Sample Bill To Patient INSTITUTIONAL BILLING Institution Name Institution Code Institution Contact Name Institution Phone Institution Contact Email INSURANCE Do Not Perform Test Until Patient is Aware of Out-Of-Pocket Costs (excludes prenatal testing) REQUIRED ITEMS 1. Copy of the Front/Back of Insurance Card(s) 2. ICD10 Diagnosis Code(s) 3. Name of Ordering Physician 4. Insured Signature of Authorization / / / / Name of Insured Insured Date of Birth (MM / DD / YYYY) Name of Insured Insured Date of Birth (MM / DD / YYYY) Patient's Relationship to Insured Phone of Insured Patient's Relationship to Insured Phone of Insured Address of Insured Address of Insured City State Zip City State Zip Primary Insurance Co. Name Primary Insurance Co. Phone Secondary Insurance Co. Name Secondary Insurance Co. Phone Primary Member Policy # Primary Member Group # Secondary Member Policy # Secondary Member Group # By signing below, I hereby authorize Baylor Genetics to provide my insurance carrier any information necessary, including test results, for processing my insurance claim. -

EUROPEAN PATENT OFFICE, VIENNA Thousand Oaks, CA 91320 (US) SUB-OFFICE
Europäisches Patentamt *EP001033405A2* (19) European Patent Office Office européen des brevets (11) EP 1 033 405 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.7: C12N 15/29, C12N 15/82, 06.09.2000 Bulletin 2000/36 C07K 14/415, C12Q 1/68, A01H 5/00 (21) Application number: 00301439.6 (22) Date of filing: 25.02.2000 (84) Designated Contracting States: • Brover, Vyacheslav AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU Calabasas, CA 91302 (US) MC NL PT SE • Chen, Xianfeng Designated Extension States: Los Angeles, CA 90025 (US) AL LT LV MK RO SI • Subramanian, Gopalakrishnan Moorpark, CA 93021 (US) (30) Priority: 25.02.1999 US 121825 P • Troukhan, Maxim E. 27.07.1999 US 145918 P South Pasadena, CA 91030 (US) 28.07.1999 US 145951 P • Zheng, Liansheng 02.08.1999 US 146388 P Creve Coeur, MO 63141 (US) 02.08.1999 US 146389 P • Dumas, J. 02.08.1999 US 146386 P , (US) 03.08.1999 US 147038 P 04.08.1999 US 147302 P (74) Representative: 04.08.1999 US 147204 P Bannerman, David Gardner et al More priorities on the following pages Withers & Rogers, Goldings House, (83) Declaration under Rule 28(4) EPC (expert 2 Hays Lane solution) London SE1 2HW (GB) (71) Applicant: Ceres Incorporated Remarks: Malibu, CA 90265 (US) THE COMPLETE DOCUMENT INCLUDING REFERENCE TABLES AND THE SEQUENCE (72) Inventors: LISTING IS AVAILABLE ON CD-ROM FROM THE • Alexandrov, Nickolai EUROPEAN PATENT OFFICE, VIENNA Thousand Oaks, CA 91320 (US) SUB-OFFICE. -
Supporting Information
Supporting Information: Figure S1abcd. Supplementary phylogenetic trees. Values at nodes represent support from ML bootstrap pseudoreplicates. (a) Multi-gene ML phylogeny of Tremblaya within Betaproteobacteria inferred from 49 concatenated protein sequences. (b) Zoomed-in Tremblaya ML phylogeny inferred from the 16S-23S rRNA alignment. (c) Multi-gene mealybug ML phylogeny inferred from the 419 concatenated CEGMA protein sequences. (d) ML phylogeny of gammaproteobacterial symbionts inferred from the 16S-23S rRNA alignment. Clade labels A-G were adopted from Thao et al. 2002. Figure S2ab. Schematic diagrams of insect scaffolds containing HGTs involved in amino acid and B-vitamin metabolism. Insect exons (predicted by GeneMark ES) are color-coded as green rectangles and when in close proximity to HGTs, annotated by their putative functions. Genes of bacterial origin are highlighted in yellow. (a) Genome localization of bioABD, ribAD, lysA, dapF, and tms HGTs confirming that they are present on insect scaffolds. Only the longest scaffold for each HGT is shown as the scaffolds from different mealybug species share gene order. (b) Alignments of M. hirsutus, P. marginatus and F. virgata scaffolds showing cysK acquisition after divergence of Maconellicoccus clade and cysK duplication in F. virgata (also present in P. citri and P. longispinus), and riboflavin transporter duplication in P. marginatus. Figure S3a-u. Phylogenetic trees for individual HGTs. Values at nodes represent support from ML bootstrap pseudoreplicates. Extremely short inner branches were extended by dashed lines for better legibility. Table S1ab. Supplementary tables with Tremblaya gene information. (a) Gene order, functional categories from Clusters of Orthologous groups (COG), Enzyme Commission numbers (E.C.), protein products, and gene abbreviations for all Tremblaya genomes.