Molecular and Comparative Genetics of Mental Retardation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CRASH Syndrome: Clinical Spectrum Vance Lemmon0 Guy Van Campa of Corpus Callosum Hypoplasia, Lieve Vitsa Retardation, Adducted Thumbs, Paul Couckea Patrick J
Review 1608178 Eur J Hum Genet 1995;3:273-284 Erik Fransen3' CRASH Syndrome: Clinical Spectrum Vance Lemmon0 Guy Van Campa of Corpus Callosum Hypoplasia, Lieve Vitsa Retardation, Adducted Thumbs, Paul Couckea Patrick J. Willemsa Spastic Paraparesis and a Department of Medical Genetics, Hydrocephalus Due to Mutations in University of Antwerp, Belgium; One Single Gene, L1 b Case Western Reserve University, Cleveland, Ohio, USA Keywords Abstract X-linked disorder LI is a neuronal cell adhesion molecule with important func Mental retardation tions in the development of the nervous system. The gene Hydrocephalus encoding LI is located near the telomere of the long arm of the MASA syndrome X chromosome in Xq28. We review here the evidence that Adducted thumbs several X-linked mental retardation syndromes including X- Corpus callosum agenesis linked hydrocephalus (HSAS), MASA syndrome, X-linked Spastic paraplegia complicated spastic paraparesis (SPI) and X-linked corpus LI callosum agenesis (ACC) are all due to mutations in the LI CRASH gene. The inter- and intrafamilial variability in families with Mutation analysis an LI mutation is very wide, and patients with HSAS, MASA, SPI and ACC can be present within the same family. There fore, we propose here to refer to this clinical syndrome with the acronym CRASH, for Corpus callosum hypoplasia, Retar dation, Adducted thumbs, Spastic paraplegia and Hydroceph alus. Clinical Aspects for Hydrocephalus due to Stenosis of the Aqueduct of Sylvius. This designation was X-Linked Hydrocephalus based upon the presence of aqueductal steno X-linked hydrocephalus (MIM No. sis in many HSAS patients [1]. However, later 307000) was originally described by Bickers studies reported several HSAS patients with and Adams in 1949. -

A Yeast Phenomic Model for the Gene Interaction Network Modulating
Louie et al. Genome Medicine 2012, 4:103 http://genomemedicine.com/content/4/12/103 RESEARCH Open Access A yeast phenomic model for the gene interaction network modulating CFTR-ΔF508 protein biogenesis Raymond J Louie3†, Jingyu Guo1,2†, John W Rodgers1, Rick White4, Najaf A Shah1, Silvere Pagant3, Peter Kim3, Michael Livstone5, Kara Dolinski5, Brett A McKinney6, Jeong Hong2, Eric J Sorscher2, Jennifer Bryan4, Elizabeth A Miller3* and John L Hartman IV1,2* Abstract Background: The overall influence of gene interaction in human disease is unknown. In cystic fibrosis (CF) a single allele of the cystic fibrosis transmembrane conductance regulator (CFTR-ΔF508) accounts for most of the disease. In cell models, CFTR-ΔF508 exhibits defective protein biogenesis and degradation rather than proper trafficking to the plasma membrane where CFTR normally functions. Numerous genes function in the biogenesis of CFTR and influence the fate of CFTR-ΔF508. However it is not known whether genetic variation in such genes contributes to disease severity in patients. Nor is there an easy way to study how numerous gene interactions involving CFTR-ΔF would manifest phenotypically. Methods: To gain insight into the function and evolutionary conservation of a gene interaction network that regulates biogenesis of a misfolded ABC transporter, we employed yeast genetics to develop a ‘phenomic’ model, in which the CFTR-ΔF508-equivalent residue of a yeast homolog is mutated (Yor1-ΔF670), and where the genome is scanned quantitatively for interaction. We first confirmed that Yor1-ΔF undergoes protein misfolding and has reduced half-life, analogous to CFTR-ΔF. Gene interaction was then assessed quantitatively by growth curves for approximately 5,000 double mutants, based on alteration in the dose response to growth inhibition by oligomycin, a toxin extruded from the cell at the plasma membrane by Yor1. -

The Purification and Identification of Interactors to Elucidate Novel Connections in the HEK 293 Cell Line
The Purification and Identification of Interactors to Elucidate Novel Connections in the HEK 293 Cell Line Brett Hawley Biochemistry, Microbiology and Immunology Faculty of Medicine University of Ottawa © Brett Hawley, Ottawa, Canada, 2012 ABSTRACT The field of proteomics studies the structure and function of proteins in a large scale and high throughput manner. My work in the field of proteomics focuses on identifying interactions between proteins and discovering novel interactions. The identification of these interactions provides new information on metabolic and disease pathways and the working proteome of a cell. Cells are lysed and purified using antibody based affinity purification followed by digestion and identification using an HPLC coupled to a mass spectrometer. In my studies, I looked at the interaction networks of several AD related genes (Apolipoprotein E, Clusterin variant 1 and 2, Low-density lipoprotein receptor, Phosphatidylinositol binding clathrin assembly protein, Alpha- synuclein and Platelet-activating factor receptor) and an endosomal recycling pathway involved in cholesterol metabolism (Eps15 homology domain 1,2 and 4, Proprotein convertase subtilisin/kexin type 9 and Low-density lipoprotein receptor). Several novel and existing interactors were identified and these interactions were validated using co-immunopurification, which could be the basis for future research. ii ACKNOWLEDGEMENTS I would like to take this opportunity to thank my supervisor, Dr. Daniel Figeys, for his support and guidance throughout my studies in his lab. It was a great experience to work in his lab and I am very thankful I was given the chance to learn and work under him. I would also like to thank the members of my lab for all their assistance in learning new techniques and equipment in the lab. -

The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination
The Journal of Neuroscience, August 31, 2011 • 31(35):12579–12592 • 12579 Cellular/Molecular The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination Junji Yamauchi,1,3,5 Yuki Miyamoto,1 Hajime Hamasaki,1,3 Atsushi Sanbe,1 Shinji Kusakawa,1 Akane Nakamura,2 Hideki Tsumura,2 Masahiro Maeda,4 Noriko Nemoto,6 Katsumasa Kawahara,5 Tomohiro Torii,1 and Akito Tanoue1 1Department of Pharmacology and 2Laboratory Animal Resource Facility, National Research Institute for Child Health and Development, Setagaya, Tokyo 157-8535, Japan, 3Department of Biological Sciences, Tokyo Institute of Technology, Midori, Yokohama 226-8501, Japan, 4IBL, Ltd., Fujioka, Gumma 375-0005, Japan, and 5Department of Physiology and 6Bioimaging Research Center, Kitasato University School of Medicine, Sagamihara, Kanagawa 252-0374, Japan In development of the peripheral nervous system, Schwann cells proliferate, migrate, and ultimately differentiate to form myelin sheath. In all of the myelination stages, Schwann cells continuously undergo morphological changes; however, little is known about their underlying molecular mechanisms. We previously cloned the dock7 gene encoding the atypical Rho family guanine-nucleotide exchange factor (GEF) and reported the positive role of Dock7, the target Rho GTPases Rac/Cdc42, and the downstream c-Jun N-terminal kinase in Schwann cell migration (Yamauchi et al., 2008). We investigated the role of Dock7 in Schwann cell differentiation and myelination. Knockdown of Dock7 by the specific small interfering (si)RNA in primary Schwann cells promotes dibutyryl cAMP-induced morpholog- ical differentiation, indicating the negative role of Dock7 in Schwann cell differentiation. It also results in a shorter duration of activation of Rac/Cdc42 and JNK, which is the negative regulator of myelination, and the earlier activation of Rho and Rho-kinase, which is the positive regulator of myelination. -

Hereditary Spastic Paraparesis: a Review of New Developments
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.69.2.150 on 1 August 2000. Downloaded from 150 J Neurol Neurosurg Psychiatry 2000;69:150–160 REVIEW Hereditary spastic paraparesis: a review of new developments CJ McDermott, K White, K Bushby, PJ Shaw Hereditary spastic paraparesis (HSP) or the reditary spastic paraparesis will no doubt Strümpell-Lorrain syndrome is the name given provide a more useful and relevant classifi- to a heterogeneous group of inherited disorders cation. in which the main clinical feature is progressive lower limb spasticity. Before the advent of Epidemiology molecular genetic studies into these disorders, The prevalence of HSP varies in diVerent several classifications had been proposed, studies. Such variation is probably due to a based on the mode of inheritance, the age of combination of diVering diagnostic criteria, onset of symptoms, and the presence or other- variable epidemiological methodology, and wise of additional clinical features. Families geographical factors. Some studies in which with autosomal dominant, autosomal recessive, similar criteria and methods were employed and X-linked inheritance have been described. found the prevalance of HSP/100 000 to be 2.7 in Molise Italy, 4.3 in Valle d’Aosta Italy, and 10–12 Historical aspects 2.0 in Portugal. These studies employed the In 1880 Strümpell published what is consid- diagnostic criteria suggested by Harding and ered to be the first clear description of HSP.He utilised all health institutions and various reported a family in which two brothers were health care professionals in ascertaining cases aVected by spastic paraplegia. The father was from the specific region. -

Dependent Traits in Mice Models of Hyperthyroidism and Hypothyroidism
Phenotypical characterization of sex- dependent traits in mice models of hyperthyroidism and hypothyroidism Inaugural-Dissertation zur Erlangung des Doktorgrades Dr. rer. nat. der Fakultät für Biologie an der Universität Duisburg-Essen vorgelegt von Helena Rakov aus St. Petropawlowsk April 2017 Die der vorliegenden Arbeit zugrunde liegenden Experimente wurden am Universitätsklinikum Essen in der Klinik für Endokrinologie und Stoffwechselerkrankungen durchgeführt. 1. Gutachter: Prof. Dr. Dr. Dagmar Führer-Sakel 2. Gutachter: Prof. Dr. Elke Cario Vorsitzender des Prüfungsausschusses: Prof. Dr. Ruth Grümmer Tag der mündlichen Prüfung: 17.07.2017 Publications Publications Engels Kathrin*, Rakov Helena *, Zwanziger Denise, Moeller Lars C., Homuth Georg, Köhrle Josef, Brix Klaudia, Fuhrer Dagmar. Differences in mouse hepatic thyroid hormone transporter expression with age and hyperthyroidism. Eur Thyroid J 2015;4(suppl 1):81–86. DOI: 10.1159/000381020. *contributed equally Zwanziger Denise*, Rakov Helena*, Engels Kathrin, Moeller Lars C., Fuhrer Dagmar. Sex-dependent claudin-1 expression in liver of eu- and hypothyroid mice. Eur Thyroid J. 2015 Sep; 4(Suppl 1): 67–73. DOI: 10.1159/000431316. *contributed equally Engels Kathrin*, Rakov Helena*, Zwanziger Denise, Hoenes Georg Sebastian, Rehders Maren, Brix Klaudia, Koehrle Josef, Moeller Lars Christian, Fuhrer Dagmar. Efficacy of protocols for induction of chronic hyperthyroidism in male and female mice. Endocrine. 2016 Oct;54(1):47-54. DOI: 10.1007/s12020-016-1020-8. Rakov Helena*, Engels Kathrin*, Hönes Georg Sebastian, Strucksberg Karl-Heinz, Moeller Lars Christian, Köhrle Josef, Zwanziger Denise, Führer Dagmar. Sex-specific phenotypes of hyperthyroidism and hypothyroidism in mice. Biol Sex Differ. 2016 Aug 24;7(1):36. DOI: 10.1186/s13293-016-0089-3. -

Discovery and the Genic Map of the Human Genome
Downloaded from genome.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press RESEARCH The Genexpress Index: A Resource for Gene Discovery and the Genic Map of the Human Genome R6mi Houlgatte, 1'2'3' R6gine Mariage-Samson, 1'2'3 Simone Duprat, 2 Anne Tessier, 2 Simone Bentolila, 1'2 Bernard Lamy, 2 and Charles Auffray 1'2'4 1Genexpress, Centre National de la Recherche Scientifique (CNRS) UPR420, 94801 Villejuif CEDEX, France; 2Genexpress, G4n6thon, 91002 Evry CEDEX, France Detailed analysis of a set of 18,698 sequences derived from both ends of 10,979 human skeletal muscle and brain cDNA clones defined 6676 functional families, characterized by their sequence signatures over 5750 distinct human gene transcripts. About half of these genes have been assigned to specific chromosomes utilizing 2733 eSTS markers, the polymerase chain reaction, and DNA from human-rodent somatic cell hybrids. Sequence and clone clustering and a functional classification together with comprehensive data base searches and annotations made it possible to develop extensive sequence and map cross-indexes, define electronic expression profiles, identify a new set of overlapping genes, and provide numerous new candidate genes for human pathologies. During the last 20 years, since the first descrip- 1993; Park et al. 1993; Takeda et al. 1993; Affara tions of eucaryotic cDNA cloning (Rougeon et al. et al. 1994; Davies et al. 1994; Kerr et al. 1994; 1975; Efstratiadis et al. 1976), cDNA studies have Konishi et al. 1994; Kurata et al. 1994; Liew et al. played a central role in molecular genetics. Early 1994; Murakawa et al. -
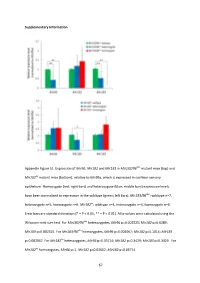
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

DNMT-Focused Library
DNMT-focused library Medicinal and Computational Chemistry Dept., ChemDiv, Inc., 6605 Nancy Ridge Drive, San Diego, CA 92121 USA, Service: +1 877 ChemDiv, Tel: +1 858-794-4860, Fax: +1 858-794-4931, Email: [email protected] DNA methylation is an essential intracellular event critically involved in a wide range of endogenous processes that play significant role in the foundation of genetic phenomena and diseases. Such epigenetic modifications play a key role in the patho-physiology of many tumors and the current use of agents targeting epigenetic changes has become a topic of intense interest in cancer research. Particularly, DNA methyltransferase (DNMT) is a crucial enzyme for cytosine methylation in DNA [1]. In mammals, DNMTs can be broadly divided into two functional families (DNMT1 and DNMT3). All mammalian DNA methyltransferases are encoded by their own single gene, and consisted of catalytic and regulatory regions (except DNMT2). Via interactions between functional domains in the regulatory or catalytic regions and other adaptors or cofactors, DNA methyltransferases can be localized at selective areas (specific DNA/nucleotide sequence) and linked to specific chromosome status (euchromatin/heterochromatin, various histone modification status). With assistance from UHRF1 and DNMT3L or other factors in DNMT1 and DNMT3a/ DNMT3b, mammalian DNA methyltransferases can be recruited, and then specifically bind to hemimethylated and unmethylated double-stranded DNA sequence to maintain and de novo setup patterns for DNA methylation. Complicated -

Mechanisms for the Inhibition of DNA Methyltransferases by Tea Catechins and Bioflavonoids
Molecular Pharmacology Fast Forward. Published on July 21, 2005 as DOI: 10.1124/mol.104.008367 Molecular PharmacologyThis article has Fastnot been Forward. copyedited Publishedand formatted. Theon finalJuly version 21, 2005 may differ as doi:10.1124/mol.104.008367from this version. MOL 8367 Mechanisms for the Inhibition of DNA Methyltransferases by Tea Catechins and Bioflavonoids Downloaded from Won Jun Lee, Joong-Youn Shim and Bao Ting Zhu1 Department of Basic Pharmaceutical Sciences, College of Pharmacy, University of South Carolina, Columbia, SC 29208, USA (W.J.L., B.T.Z.) molpharm.aspetjournals.org and J. L. Chambers Biomedical/Biotechnology Research Institute, North Carolina Central University, Durham, NC 27707 (J.-Y.S.) at ASPET Journals on September 27, 2021 1 Copyright 2005 by the American Society for Pharmacology and Experimental Therapeutics. Molecular Pharmacology Fast Forward. Published on July 21, 2005 as DOI: 10.1124/mol.104.008367 This article has not been copyedited and formatted. The final version may differ from this version. MOL 8367 Running title: Modulation of DNA methylation by dietary polyphenols Corresponding author: Bao Ting Zhu, Ph.D. Frank and Josie P. Fletcher Professor of Pharmacology and an American Cancer Society Research Scholar, and to whom requests for reprints should be addressed at the Department of Basic Pharmaceutical Sciences, College of Pharmacy, University of South Carolina, Room 617 of Coker Life Sciences Building, 700 Sumter Street, Columbia, SC 29208 (USA). PHONE: 803-777-4802; FAX: 803-777-8356; E-MAIL: [email protected] Number of text pages : 34 Downloaded from Number of tables : 2 Number of figures : 10 Number of references : 39 molpharm.aspetjournals.org Number of words in abstract : 229 Number of words in introduction : 458 Number of words in discussion : 1567 at ASPET Journals on September 27, 2021 2 Molecular Pharmacology Fast Forward. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Orphanet Report Series Rare Diseases Collection
Marche des Maladies Rares – Alliance Maladies Rares Orphanet Report Series Rare Diseases collection DecemberOctober 2013 2009 List of rare diseases and synonyms Listed in alphabetical order www.orpha.net 20102206 Rare diseases listed in alphabetical order ORPHA ORPHA ORPHA Disease name Disease name Disease name Number Number Number 289157 1-alpha-hydroxylase deficiency 309127 3-hydroxyacyl-CoA dehydrogenase 228384 5q14.3 microdeletion syndrome deficiency 293948 1p21.3 microdeletion syndrome 314655 5q31.3 microdeletion syndrome 939 3-hydroxyisobutyric aciduria 1606 1p36 deletion syndrome 228415 5q35 microduplication syndrome 2616 3M syndrome 250989 1q21.1 microdeletion syndrome 96125 6p subtelomeric deletion syndrome 2616 3-M syndrome 250994 1q21.1 microduplication syndrome 251046 6p22 microdeletion syndrome 293843 3MC syndrome 250999 1q41q42 microdeletion syndrome 96125 6p25 microdeletion syndrome 6 3-methylcrotonylglycinuria 250999 1q41-q42 microdeletion syndrome 99135 6-phosphogluconate dehydrogenase 67046 3-methylglutaconic aciduria type 1 deficiency 238769 1q44 microdeletion syndrome 111 3-methylglutaconic aciduria type 2 13 6-pyruvoyl-tetrahydropterin synthase 976 2,8 dihydroxyadenine urolithiasis deficiency 67047 3-methylglutaconic aciduria type 3 869 2A syndrome 75857 6q terminal deletion 67048 3-methylglutaconic aciduria type 4 79154 2-aminoadipic 2-oxoadipic aciduria 171829 6q16 deletion syndrome 66634 3-methylglutaconic aciduria type 5 19 2-hydroxyglutaric acidemia 251056 6q25 microdeletion syndrome 352328 3-methylglutaconic