Investigating the Molecular Signatures of Β1-Adrenergic Receptor Activation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Adrenergic Antagonist
PHARMACOLOGY Adrenergic antagonist OBJECTIVES: • Describe the different classifications for drugs that can block sympathetic nervous system. •Describe the kinetics, dynamics, uses and side effects of alpha adrenergic drugs. • Identify Difference between selective and non selective alpha blockers. • Know the difference between tamsulosin and other selective alpha receptor blockers. •Identify the different classifications for beta receptors blockers. •Describe the kinetics, dynamics, uses and side effects of beta adrenergic drugs. •Know the preferable drug for diseases as hypertension, glaucoma, arrythmia, myocardial infarction, anxiety, migraine and ect…. • Important. • Extra notes It’s a recall, if you know it you can skip it! Adrenergic receptors Adrenergic receptors Dopaminergic adrenoceptors adrenoceptors α− β− receptors β3 α1 α2 β1 β2 e.g. D1 α1 β2 β1 β3 Post-synaptic excitatory in function (cause inhibitory in function excitatory in In adipose contraction) except in GIT. (cause relaxation) function, present tissue mainly in heart Present mainly in smooth muscles. Contraction of pregnant Relaxation of the uterus ↑ heart rate: ↑ lipolysis uterus. (Delay premature labor) + chronotropic ↑ free fatty effect, Vasoconstriction of skin & Relaxation of skeletal & acids. Tachycardia peripheral blood vessels coronary blood vessels →increased peripheral (vasodilatation) ↑ force of → resistance hypertension. contraction : Relaxation of GIT muscles & urinary bladder’s muscles. + inotropic effect Contraction of GIT sphincter (constipation) & urinary -

Guanfacine Extended Release for ADHD
Out of the Pipeline p Guanfacine extended release for ADHD Floyd R. Sallee, MD, PhD uanfacine extended release (GXR)— Table 1 α Once-daily a selective -2 adrenergic agonist Guanfacine extended release: GFDA-approved for the treatment formulation may of attention-defi cit/hyperactivity disor- Fast facts improve adherence der (ADHD)—has demonstrated effi cacy Brand name: Intuniv and control for inattentive and hyperactive/impulsive Indication: Attention-defi cit/hyperactivity symptoms across disorder symptom domains in 2 large trials lasting® Dowden Health Media a full day 8 and 9 weeks.1,2 GXR’s once-daily formu- Approval date: September 3, 2009 lation may increase adherence and deliver Availability date: November 2009 consistent control of symptomsCopyright across a For personalManufacturer: use Shire only full day (Table 1). Dosing forms: 1-mg, 2-mg, 3-mg, and 4-mg extended-release tablets Recommended dosage: 0.05 to 0.12 mg/kg Clinical implications once daily GXR exhibits enhancement of noradren- ergic pathways through selective direct receptor action in the prefrontal cortex.3 brain believed to play a major role in at- This mechanism of action is different from tentional and organizational functions that that of other FDA-approved ADHD medi- preclinical research has linked to ADHD.3 cations. GXR can be used alone or in com- The postsynaptic α-2A receptor is bination with stimulants or atomoxetine thought to play a central role in the opti- for treating complex ADHD, such as cases mal functioning of the PFC as illustrated accompanied by oppositional features and by the “inverted U hypothesis of PFC ac- emotional dysregulation or characterized tivation.”4 In this model, cyclic adenos- by partial stimulant response. -

The Adrenergic Control of Lower Esophageal Sphincter Function: an EXPERIMENTAL MODEL of DENERVATION SUPERSENSITIVITY
The Adrenergic Control of Lower Esophageal Sphincter Function: AN EXPERIMENTAL MODEL OF DENERVATION SUPERSENSITIVITY Anthony J. DiMarino, Sidney Cohen J Clin Invest. 1973;52(9):2264-2271. https://doi.org/10.1172/JCI107413. To evaluate the adrenergic regulation of lower esophageal sphincter (LES) function, LES pressure, LES relaxation during swallowing, and blood pressure were measured in the anesthetized opossum, Didelphis virginiana, during intravenous administration of alpha and beta adrenergic agonists and antagonists. Studies were done in controls and animals adrenergically denervated with 6-hydroxydopamine. Alpha adrenergic agonists (norepinephrine, phenylephrine) increased LES pressure and blood pressure, whereas a beta adrenergic agonist (isoproterenol) decreased both pressures. Alpha adrenergic antagonism (phentolamine) reduced basal LES pressure by 38.3±3.8% (mean ±SEM) (P < 0.001). Beta adrenergic antagonism (propranolol) had no significant effect on either basal LES pressure or percent of LES relaxation with swallowing. After adrenergic denervation with 6-hydroxydopamine, basal LES pressure was reduced by 22.5±5.3% (P < 0.025) but LES relaxation during swallowing was unaltered. In denervated animals, both LES pressure and blood pressure dose response curves showed characteristics of denervation supersensitivity to alpha but not to beta adrenergic agonists. These studies suggest: (a) a significant portion of basal LES pressure is dependent upon alpha adrenergic stimulation; (b) LES relaxation during swallowing is not an adrenergically mediated response; c( ) the LES pressure response to alpha adrenergic agonists after 6-hydroxydopamine may serve as a model of denervation supersensitivity in the gastrointestinal tract. Find the latest version: https://jci.me/107413/pdf The Adrenergic Control of Lower Esophageal Sphincter Function AN EXPERIMENTAL MODEL OF DENERVATION SUPERSENSITIVITY ANTHoNY J. -

The Importance of Serotonergic and Adrenergic Receptors for the Induction and Expression of One-Trial Cocaine-Induced Behavioral Sensitization" (2016)
California State University, San Bernardino CSUSB ScholarWorks Electronic Theses, Projects, and Dissertations Office of aduateGr Studies 12-2016 THE IMPORTANCE OF SEROTONERGIC AND ADRENERGIC RECEPTORS FOR THE INDUCTION AND EXPRESSION OF ONE- TRIAL COCAINE-INDUCED BEHAVIORAL SENSITIZATION Krista N. Rudberg California State University - San Bernardino Follow this and additional works at: https://scholarworks.lib.csusb.edu/etd Part of the Biological Psychology Commons, and the Pharmacology Commons Recommended Citation Rudberg, Krista N., "THE IMPORTANCE OF SEROTONERGIC AND ADRENERGIC RECEPTORS FOR THE INDUCTION AND EXPRESSION OF ONE-TRIAL COCAINE-INDUCED BEHAVIORAL SENSITIZATION" (2016). Electronic Theses, Projects, and Dissertations. 420. https://scholarworks.lib.csusb.edu/etd/420 This Thesis is brought to you for free and open access by the Office of aduateGr Studies at CSUSB ScholarWorks. It has been accepted for inclusion in Electronic Theses, Projects, and Dissertations by an authorized administrator of CSUSB ScholarWorks. For more information, please contact [email protected]. THE IMPORTANCE OF SEROTONERGIC AND ADRENERGIC RECEPTORS FOR THE INDUCTION AND EXPRESSION OF ONE-TRIAL COCAINE- INDUCED BEHAVIORAL SENSITIZATION A Thesis Presented to the Faculty of California State University, San Bernardino In Partial Fulfillment of the Requirements for the Degree Master of Arts in General/Experimental Psychology by Krista Nicole Rudberg December 2016 THE IMPORTANCE OF SEROTONERGIC AND ADRENERGIC RECEPTORS FOR THE INDUCTION AND EXPRESSION OF ONE-TRIAL COCAINE- INDUCED BEHAVIORAL SENSITIZATION A Thesis Presented to the Faculty of California State University, San Bernardino by Krista Nicole Rudberg December 2016 Approved by: Sanders McDougall, Committee Chair, Psychology Cynthia Crawford, Committee Member Matthew Riggs, Committee Member © 2016 Krista Nicole Rudberg ABSTRACT Addiction is a complex process in which behavioral sensitization may be an important component. -

F3-Adrenergic Antagonist (Receptors/Cyclic AMP/Aminobenzylpropranolol/Iodohydroxybenzylpindolol/Isoproterenol) WESLEY L
Proc. Natl. Acad. Sci. USA Vol. 76, No. 12, pp. 6401-6405, December 1979 Cell Biology Quantitative relationship between 3-adrenergic receptor number and physiologic responses as studied with a long-lasting f3-adrenergic antagonist (receptors/cyclic AMP/aminobenzylpropranolol/iodohydroxybenzylpindolol/isoproterenol) WESLEY L. TERASAKI, JOEL LINDEN, AND GARY BROOKER* Department of Pharmacology, School of Medicine, University of Virginia, Charlottesville, Virginia 22908 Communicated by Paul Greengard, August 20, 1979 ABSTRACT The aminobenzyl analog of propranolol, atrium when f3-adrenergic receptor number is selectively de- 1- (p-amino-a,a-dimethylphenethylamino)-3(1-naphthoxy)2- creased. To investigate this problem, a new propanol, was synthesized and found tobe a potent B-adrenergic f3-adrenergic blocking agent. The fl-adrenergic receptors of cultured rat C6 blocking agent, 1-(p-amino-a,a-dimethylphenethylamino- glioma cells (2B clone) as assessed by [' 51]iodohydroxybenzyl- 3-(1-naphthoxy)-2-propanol (aminobenzylpropranolol), was pindolol binding were decreased 50 and >95% afterpretreat- synthesized. It was found to be a selective agent for the elimi- ment with 8 nM and 1 ,M aminobenzylpropranolol, respec- nation of cellular /3-adrenergic receptors. tively. Unlike propranolol, aminobenzylpropranolol displayed a prolonged blockade of receptors that was maintained during several hours of washing. [121s]Iodohydroxybenzylpindolol MATERIALS AND METHODS saturation binding experiments in cells exposed to aminoben- Tissue Culture. C6 rat glioma cells, 2B subclone (7), were zylpropranolol and subsequently washed indicated that the compound effectively diminished receptor number with no grown in monolayer cultures as described (1) in 16-mm plastic change in the affinity of the remaining receptors for iodohy- cluster dishes. In all experiments, the cells were changed from droxybenzylpindolol. -

Mutation of the 2A-Adrenoceptor Impairs Working Memory Performance and Annuls Cognitive Enhancement by Guanfacine
The Journal of Neuroscience, October 1, 2002, 22(19):8771–8777 ␣ Mutation of the 2A-Adrenoceptor Impairs Working Memory Performance and Annuls Cognitive Enhancement by Guanfacine Jenna S. Franowicz,1 Lynn E. Kessler,1 Catherine M. Dailey Borja,1 Brian K. Kobilka,2 Lee E. Limbird,3 and Amy F. T. Arnsten1 1Department of Neurobiology, Yale University School of Medicine, New Haven, Connecticut 06510, 2Howard Hughes Medical Institute and Departments of Medicine and Molecular and Cellular Physiology, Stanford University, Palo Alto, California 94305, and 3Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, Tennessee 37232 Norepinephrine strengthens the working memory, behavioral cortical damage or a spatial discrimination control task with inhibition, and attentional functions of the prefrontal cortex similar motor and motivational demands but no dependence on ␣ ␣ through actions at postsynaptic 2-adrenoceptors ( 2-AR). The prefrontal cortex. The effects of guanfacine on performance of ␣ 2-AR agonist guanfacine enhances prefrontal cortical func- the delayed alternation task were assessed in additional groups ␣ tions in rats, monkeys, and human beings and ameliorates of wild-type versus 2A-AR mutant mice. We observed that ␣ prefrontal cortical deficits in patients with attention deficit hy- functional loss of the 2A-AR subtype, unlike knock-out of the ␣ peractivity disorder. The present study examined the subtype of 2C-AR subtype, weakened performance of the prefrontal cor- ␣ 2-AR underlying these beneficial effects. Because there are no tical task without affecting learning and resulted in loss of the ␣ ␣ ␣ selective 2A-AR, 2B-AR, or 2C-AR agonists or antagonists, beneficial response to guanfacine. These data demonstrate the ␣ genetically altered mice were used to identify the molecular importance of 2A-AR subtype stimulation for the cognitive target of the action of guanfacine. -

Synergistic Action of 5-HT2A Antagonists and Selective Serotonin Reuptake Inhibitors in Neuropsychiatric Disorders
Neuropsychopharmacology (2003) 28, 402–412 & 2003 Nature Publishing Group All rights reserved 0893-133X/03 $25.00 www.neuropsychopharmacology.org Synergistic Action of 5-HT2A Antagonists and Selective Serotonin Reuptake Inhibitors in Neuropsychiatric Disorders ,1 2 3 2 Gerard J Marek* , Linda L Carpenter , Christopher J McDougle and Lawrence H Price 1Department of Psychiatry, Yale School of Medicine, New Haven, CT, USA; 2Department of Psychiatry and Human, Brown Medical School, Mood 3 Disorders Program, Behavior, Butler Hospital, Providence, RI, USA; Department of Psychiatry, Indiana University School of Medicine, Indianapolis, IN, USA Recently, the addition of drugs with prominent 5-HT2 receptor antagonist properties (risperidone, olanzapine, mirtazapine, and mianserin) to selective serotonin reuptake inhibitors (SSRIs) has been shown to enhance therapeutic responses in patients with major depression and treatment-refractory obsessive–compulsive disorder (OCD). These 5-HT antagonists may also be effective in 2 ameliorating some symptoms associated with autism and other pervasive developmental disorders (PDDs). At the doses used, these drugs would be expected to saturate 5-HT2A receptors. These findings suggest that the simultaneous blockade of 5-HT2A receptors and activation of an unknown constellation of other 5-HT receptors indirectly as a result of 5-HT uptake inhibition might have greater therapeutic efficacy than either action alone. Animal studies have suggested that activation of 5-HT1A and 5-HT2C receptors may counteract the effects of activating 5-HT2A receptors. Additional 5-HT receptors, such as the 5-HT1B/1D/5/7 receptors, may similarly counteract the effects of 5-HT receptor activation. These clinical and preclinical observations suggest that the combination of highly 2A selective 5-HT antagonists and SSRIs, as well as strategies to combine high-potency 5-HT receptor and 5-HT transporter blockade in 2A 2A a single compound, offer the potential for therapeutic advances in a number of neuropsychiatric disorders. -

Study of Methyl Dopa Versus Labetalol in Management of Preeclampsia and Gestational Hypertension
logy & Ob o st ec e tr n i y c s G Dharwadkar et al., Gynecol Obstet (Sunnyvale) 2014, 4:9 Gynecology & Obstetrics DOI; 10.4172/2161-0932.1000242 ISSN: 2161-0932 Research Article Open Access Study of Methyl Dopa Versus Labetalol in Management of Preeclampsia and Gestational Hypertension Dharwadkar MN1, Kanakamma MK1, Dharwadkar SN2*, Rajagopal K1, Gopakumar C3, Divya James Fenn J4 and Balachandar V3,5 1Department of Obstetrics and Gynaecology, Yenepoya Medical College and teaching Hospital, Yenepoya University, Deralakatte, Mangalore, Karnataka 575022, India 2Department of Zoology, K.L.E’s. S. Nijalingappa College, KLE Medical University (Health), Bangalore, Karnataka 560010, India 3Department of Human Genetics Molecular Biology, Bharathiar University, Coimbatore, India 4Post Graduate Student, Department of Obstetrics and Gynaecology, Sree Balaji Medical College and Hospital, Chennai 600044, Tamil Nadu, India 5Human Molecular Genetics Laboratory, Department of Zoology, Bharathiar University, Coimbatore, India Abstract Objective: To assess the efficacy and safety of labetalol compared with methyldopa in the management of mild and moderate cases of pregnancy-induced hypertension (PIH). Methods: Eighty patients with PIH were randomly allocated to receive either labetalol (group A) or methyldopa (group B). Administration of drugs with respect to Age, Gravid Status, Blood Pressure, Urine albumin Levels, Side Effects, Drug dosage, Additional Treatment, Prolongation of Pregnancy, New born Screening Test (NST), mode of termination, Indication of caesarean section, Perinatal safety and APGAR scores were studied. The statistical level of significance was taken at P<0.05. Results: A labetalol has been very effective in control as well as earlier onset of action in patients with methyl dopa. -

Oral Phentolamine: an Alpha-1, Alpha-2 Adrenergic Antagonist for the Treatment of Erectile Dysfunction
International Journal of Impotence Research (2000) 12, Suppl 1, S75±S80 ß 2000 Macmillan Publishers Ltd All rights reserved 0955-9930/00 $15.00 www.nature.com/ijir Oral phentolamine: an alpha-1, alpha-2 adrenergic antagonist for the treatment of erectile dysfunction I Goldstein1 1Department of Urology, Boston University School of Medicine, Boston, MA, USA Phentolamine mesylate is an alpha-1 and alpha-2 selective adrenergic receptor antagonist which has undergone clinical trials for erectile dysfunction treatment. Biochemical and physiological studies in human erectile tissue have revealed a high af®nity of phentolamine for alpha-1 and alpha-2 adrenergic receptors. Based on pharmacokinetic studies, it is suggested that 30±40 min following oral ingestion of 40 or 80 mg of phentolamine (Vasomax), the mean plasma phentolamine concentrations are suf®cient to occupy the alpha-1 and -2 adrenergic receptors in erectile tissue and thereby result in inhibition of adrenergic-mediated physiologic activity. In large multi-center, placebo-controlled pivotal phase III clinical trials, the mean change in the erectile function domain of the International Index of Erectile Function scores (Questions 1±5 and 15) from screening to the end of treatment was signi®cantly higher following use of active drug (40 mg and 80 mg) compared to placebo. Three to four times as many patients receiving phentolamine reported being satis®ed or very satis®ed compared with those receiving placebo. At doses of 40 mg and 80 mg respectively, 55% and 59% of men were able to achieve vaginal penetration with 51% and 53% achieving penetration on 75% of attempts. -
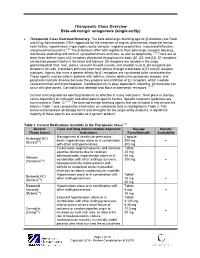
Therapeutic Class Overview Beta-Adrenergic Antagonists (Single-Entity)
Therapeutic Class Overview Beta-adrenergic antagonists (single-entity) · Therapeutic Class Overview/Summary: The beta-adrenergic blocking agents (β-blockers) are Food and Drug Administration (FDA)-approved for the treatment of angina, arrhythmias, essential tremor, heart failure, hypertension, hypertrophic aortic stenosis, migraine prophylaxis, myocardial infarction, and pheochromocytoma.1-26 The β-blockers differ with regards to their adrenergic-receptor blocking, membrane stabilizing and intrinsic sympathomimetic activities, as well as lipophilicity.1-26 There are at least three distinct types of β receptors distributed throughout the body (β1, β2, and β3). β1-receptors are located predominantly in the heart and kidneys. β2-receptors are located in the lungs, gastrointestinal tract, liver, uterus, vascular smooth muscle, and skeletal muscle. β3-receptors are located in fat cells. β-blockers primarily exert their effects through a blockade of β1 and β2 receptor subtypes. Agents that have a greater affinity for β1 receptors are considered to be cardioselective. These agents may be safer in patients with asthma, chronic obstructive pulmonary disease, and peripheral vascular disease because they produce less inhibition of β2 receptors, which mediate vasoconstriction and bronchospasm. Cardioselectivity is dose dependent; therefore, β2 blockade can occur at higher doses. Carvedilol and labetalol also block α-adrenergic receptors. 27-28 Current clinical guidelines identify β-blockers as effective in many indications. Their place in therapy varies depending on indication and other patient specific factors. Specific treatment guidelines are summarized in Table 12.29-61 The beta-adrenergic blocking agents that are included in this review are listed in Table 1 and comparative information on cardioselectivity is highlighted in Table 2. -

Long-Term Antidepressant Treatments Result in a Tonic Activation of Forebrain 5-HT1A Receptors
The Journal of Neuroscience, December 1, 1998, 18(23):10150–10156 Long-Term Antidepressant Treatments Result in a Tonic Activation of Forebrain 5-HT1A Receptors Nasser Haddjeri, Pierre Blier, and Claude de Montigny Neurobiological Psychiatry Unit, McGill University, Montre´ al, Que´ bec, Canada H3A 1A1 We report here the first direct functional evidence of an increase ministration, WAY 100635 markedly increased (60–200%) the in the tonic activation of postsynaptic 5-HT1A receptors by firing activity of CA3 pyramidal neurons. Such a disinhibition antidepressant treatments. Because 5-HT1A receptor activation was absent in rats treated with the nonantidepressant drug hyperpolarizes and inhibits CA3 pyramidal neurons in the dorsal chlorpromazine, in rats receiving only one ECS, or in rats hippocampus, we determined, using in vivo extracellular re- receiving multiple ECSs in combination with an intrahippocam- cording, whether the selective 5-HT1A receptor antagonist WAY pal pertussis toxin treatment to inactivate Gi/o-coupled 5-HT1A 100635 could disinhibit these neurons. Unexpectedly, no receptors. These data indicate that such antidepressant treat- disinhibition could be detected in controls. However, after ments, acting on entirely different primary targets, might allevi- long-term treatment with the tricyclic antidepressant imipra- ate depression by enhancing the tonic activation of forebrain mine, the selective 5-HT reuptake inhibitor paroxetine, the postsynaptic 5-HT1A receptors. a reversible monoamine oxidase-A inhibitor befloxatone, the 2- adrenergic antagonist mirtazapine, or the 5-HT1A receptor ag- Key words: antidepressants; serotonin (5-HT); 5-HT1A recep- onist gepirone or multiple electroconvulsive shock (ECS) ad- tors; WAY 100635; disinhibition; dorsal hippocampus a With a prevalence of at least 4% of the general population, major 2-adrenoceptor antagonist, increases 5-HT neurotransmission as depression is one of the most common psychiatric disorders. -

CHRONIC EFFECTS of a MONOAMINE OXIDASE-INHIBITING ANTIDEPRESSANT: DECREASES in FUNCTIONAL A-ADRENERGIC AUTORECEPTORSPRECEDETHEDE
0270-6474/82/0211-1588$02.00/O The Journal of Neuroscience Copyright 0 Society for Neuroscience Vol. 2, No. 11, pp. 1588-1595 Printed in U.S.A. November 1982 CHRONIC EFFECTS OF A MONOAMINE OXIDASE-INHIBITING ANTIDEPRESSANT: DECREASES IN FUNCTIONAL a-ADRENERGIC AUTORECEPTORSPRECEDETHEDECREASEIN NOREPINEPHRINE-STIMULATED CYCLIC ADENOSINE 3’:WMONOPHOSPHATE SYSTEMS IN RAT BRAIN1 ROBERT M. COHEN,*,” RICHARD P. EBSTEIN,*,3 JOHN W. DALY,* AND DENNIS L. MURPHY* *Clinical Neuropharmacology Branch, National Institute of Mental Health and ‘Laboratory of Bioorganic Chemistry, National Institute of Arthritis, Metabolism and Digestive Diseases, Bethesda, Maryland 20205 Received November 2, 1981; Revised April 9, 1982; Accepted May 7, 1982 Abstract Various antidepressant drugs (monoamine oxidase inhibitors and tricyclics) enhance norepineph- rine availability and lead to adaptive changes in brain noradrenergic systems, namely, decreases in the number of p receptors and in the responsiveness of adenylate cyclase to norepinephrine stimulation. After 21 days of treatment with 1 mg/kg/day of clorgyline, an A-type-selective monoamine oxidase inhibitor, but not after 3 days, there is an increase in norepinephrine release from rat brain microsacs in response to 43 mM KC1 stimulation. Microsacs prepared from 21-day clorgyline-treated animals also show a marked decrease in the inhibition of norepinephrine release caused by the az-selective agonist clonidine. These functional changes in norepinephrine release mechanisms are accompanied by a 53% reduction in brainstem (~2receptor density as measured by [3H]clonidine binding. At the same time, despite findings of a decrease in p receptor number as determined by [3H]dihydroalprenolo1 binding data, no significant decrease in the responses of cyclic adenosine 3’:5’-monophosphate (cyclic AMP) systems to norepinephrine stimulation is observed.