63: Miscellaneous Antihypertensives and Pharmacologically Related Agents
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
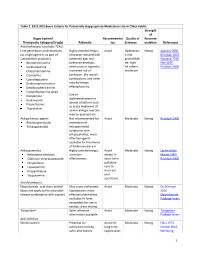
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Response to Inhaled Nitric Oxide, but Neither Sodium Nitroprusside Nor Sildenafil, Predicts Survival in Patients With
Jachec et al., J Clin Exp Cardiolog 2015, 6:6 Clinical & Experimental Cardiology http://dx.doi.org/10.4172/2155-9880.1000376 Research Article Open Access Response to Inhaled Nitric Oxide, But neither Sodium Nitroprusside nor Sildenafil, Predicts Survival in Patients with Dilated Cardiomyopathy Complicated with Pulmonary Hypertension Wojciech Jacheć1*, Celina Wojciechowska2, Andrzej Tomasik2, Damian Kawecki2, Ewa Nowalany-Kozielska2 and Jan Wodniecki2 1II Katedra i Oddział Kliniczny Kardiologii w Zabrzu Śląskiego Uniwersytetu Medycznego w Katowicach, ul. Skłodowskiej 10, 41-800 Zabrze, Polska 2Department of Cardiology in Zabrze, Medical University of Silesia in Katowice, Poland *Corresponding author: Wojciech Jacheć, II Katedra i Oddział Kliniczny Kardiologii w Zabrzu Śląskiego Uniwersytetu Medycznego w Katowicach, ul. Skłodowskiej 10, 41-800 Zabrze, Polska, Tel: +48 32 373 23 72; Fax: +48 32 271 10 10; E-mail: [email protected] Received date: May 26, 2015, Accepted date: Jun 25, 2015, Published date: Jun 29, 2015 Copyright: ©2015 Jacheć W. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Introduction: Pulmonary hypertension in patients with dilated cardiomyopathy is associated with higher mortality. Objectives: The aim of the study was to assess the predictive value of the vasodilator response to three different drugs, sodium nitroprusside, -

Inline-Supplementary-Material-1.Pdf
Appendix 1: STOPP/START criteria version 2 applied to the TRUST dataset Physiological system Criteria Criteria included Number (%) (The relevant () criteria for each participant were applied to the dataset and recorded in of Microsoft Office Excel ® (2013)) criteria included out of total criteria STOPP criteria Indication of medication A1. Any drug prescribed without an evidence-based clinical indication. X 1/3 (33.3) A2. Any drug prescribed beyond the recommended duration, where treatment duration is X well defined. A3. Any duplicate drug class prescription e.g. two concurrent NSAIDs, SSRIs, loop diuretics, ACE inhibitors, anticoagulants (optimisation of monotherapy within a single drug class should be observed prior to considering a new agent). Cardiovascular system B1. Digoxin for heart failure with preserved systolic ventricular function (no clear evidence X 7/13 (53.8) of benefit). B2. Verapamil or diltiazem with NYHA Class III or IV heart failure (may worsen heart failure). B3. Beta-blocker in combination with verapamil or diltiazem (risk of heart block). B4. Beta blocker with symptomatic bradycardia (< 50/min), type II heart block or complete heart block (risk of profound hypotension, asystole). B5. Amiodarone as first-line antiarrhythmic therapy in supraventricular tachyarrhythmias X (higher risk of side-effects than beta-blockers, digoxin, verapamil or diltiazem). B6. Loop diuretic as first-line treatment for hypertension (safer, more effective alternatives available). B7. Loop diuretic for dependent ankle oedema without clinical, biochemical evidence or radiological evidence of heart failure, liver failure, nephrotic syndrome or renal failure (leg elevation and /or compression hosiery usually more appropriate). B8. Thiazide diuretic with current significant hypokalaemia (i.e. -

Imidazoline Antihypertensive Drugs: Selective I1-Imidazoline Receptors Activation K
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by FarFar - Repository of the Faculty of Pharmacy, University of Belgrade REVIEW Imidazoline Antihypertensive Drugs: Selective I1-Imidazoline Receptors Activation K. Nikolic & D. Agbaba Faculty of Pharmacy, Institute of Pharmaceutical Chemistry, University of Belgrade, Vojvode Stepe, Belgrade, Serbia Keywords SUMMARY α2-Adrenergic receptors; Centrally acting antihypertensives; Clonidine; Hypertension; Involvement of imidazoline receptors (IR) in the regulation of vasomotor tone as well as in Imidazoline receptors; Rilmenidine. the mechanism of action of some centrally acting antihypertensives has received tremen- dous attention. To date, pharmacological studies have allowed the characterization of three Correspondence main imidazoline receptor classes, the I1-imidazoline receptor which is involved in central K. Nikolic, Faculty of Pharmacy, Institute of inhibition of sympathetic tone to lower blood pressure, the I2-imidazoline receptor which Pharmaceutical Chemistry, University of is an allosteric binding site of monoamine oxidase B (MAO-B), and the I3-imidazoline re- Belgrade, Vojvode Stepe 450, 11000 Belgrade, ceptor which regulates insulin secretion from pancreatic β-cells. All three imidazoline re- Serbia. ceptors represent important targets for cardiovascular research. The hypotensive effect of + Tel: 381-63-84-30-677; clonidine-like centrally acting antihypertensives was attributed both to α2-adrenergic re- + Fax: 381-11-3974-349; ceptors and nonadrenergic I1-imidazoline receptors, whereas their sedative action involves E-mail: [email protected] activation of only α2-adrenergic receptors located in the locus coeruleus. Since more selec- tive I1-imidazoline receptors ligands reduced incidence of typical side effects of other cen- trally acting antihypertensives, there is significant interest in developing new agents with higher selectivity and affinity for I1-imidazoline receptors. -

Adrenergic Antagonist
PHARMACOLOGY Adrenergic antagonist OBJECTIVES: • Describe the different classifications for drugs that can block sympathetic nervous system. •Describe the kinetics, dynamics, uses and side effects of alpha adrenergic drugs. • Identify Difference between selective and non selective alpha blockers. • Know the difference between tamsulosin and other selective alpha receptor blockers. •Identify the different classifications for beta receptors blockers. •Describe the kinetics, dynamics, uses and side effects of beta adrenergic drugs. •Know the preferable drug for diseases as hypertension, glaucoma, arrythmia, myocardial infarction, anxiety, migraine and ect…. • Important. • Extra notes It’s a recall, if you know it you can skip it! Adrenergic receptors Adrenergic receptors Dopaminergic adrenoceptors adrenoceptors α− β− receptors β3 α1 α2 β1 β2 e.g. D1 α1 β2 β1 β3 Post-synaptic excitatory in function (cause inhibitory in function excitatory in In adipose contraction) except in GIT. (cause relaxation) function, present tissue mainly in heart Present mainly in smooth muscles. Contraction of pregnant Relaxation of the uterus ↑ heart rate: ↑ lipolysis uterus. (Delay premature labor) + chronotropic ↑ free fatty effect, Vasoconstriction of skin & Relaxation of skeletal & acids. Tachycardia peripheral blood vessels coronary blood vessels →increased peripheral (vasodilatation) ↑ force of → resistance hypertension. contraction : Relaxation of GIT muscles & urinary bladder’s muscles. + inotropic effect Contraction of GIT sphincter (constipation) & urinary -

The In¯Uence of Medication on Erectile Function
International Journal of Impotence Research (1997) 9, 17±26 ß 1997 Stockton Press All rights reserved 0955-9930/97 $12.00 The in¯uence of medication on erectile function W Meinhardt1, RF Kropman2, P Vermeij3, AAB Lycklama aÁ Nijeholt4 and J Zwartendijk4 1Department of Urology, Netherlands Cancer Institute/Antoni van Leeuwenhoek Hospital, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands; 2Department of Urology, Leyenburg Hospital, Leyweg 275, 2545 CH The Hague, The Netherlands; 3Pharmacy; and 4Department of Urology, Leiden University Hospital, P.O. Box 9600, 2300 RC Leiden, The Netherlands Keywords: impotence; side-effect; antipsychotic; antihypertensive; physiology; erectile function Introduction stopped their antihypertensive treatment over a ®ve year period, because of side-effects on sexual function.5 In the drug registration procedures sexual Several physiological mechanisms are involved in function is not a major issue. This means that erectile function. A negative in¯uence of prescrip- knowledge of the problem is mainly dependent on tion-drugs on these mechanisms will not always case reports and the lists from side effect registries.6±8 come to the attention of the clinician, whereas a Another way of looking at the problem is drug causing priapism will rarely escape the atten- combining available data on mechanisms of action tion. of drugs with the knowledge of the physiological When erectile function is in¯uenced in a negative mechanisms involved in erectile function. The way compensation may occur. For example, age- advantage of this approach is that remedies may related penile sensory disorders may be compen- evolve from it. sated for by extra stimulation.1 Diminished in¯ux of In this paper we will discuss the subject in the blood will lead to a slower onset of the erection, but following order: may be accepted. -

Acute Effect of Sodium Nitroprusside on Microvascular Dysfunction In
Clinical Studies Acute Effect of Sodium Nitroprusside on Microvascular Dysfunction in Patients Who Underwent Percutaneous Coronary Intervention for Acute ST-segment Elevation Myocardial Infarction Kotaro Morimoto,1, 2 MD, Shigenori Ito,2 MD, Kosuke Nakasuka,2 MD, Satoru Sekimoto,2 MD, Kazuyuki Miyata,2 MD, Masahiko Inomata,2 MD, Takayuki Yoshida,2 MD, Nozomu Tamai,2 MD, Tomoaki Saeki,2 MD, Shin Suzuki,2 MD, Yoshimasa Murakami,2 MD, Koichi Sato,2 MD, Akihiro Morino,3 CE, and Yoshiyuki Shimizu,3 CE Summary Even in the era of thrombus aspiration and distal protection for ST-segment elevation acute myocardial infarction (STEMI), microvascular dysfunction does exist and improvement of microvascular dysfunction can improve the progno- sis and/or left ventricular dysfunction. We evaluated the acute effects of nitroprusside (NTP) on coronary microvascular injury that occurred after primary percutaneous coronary intervention (PCI) for STEMI in 18 patients. The final Throm- bolysis in Myocardial Infarction trial (TIMI) flow grade after PCI was 3 in 17 patients and 2 in 1 patient. The index of microcirculatory resistance (IMR) was improved significantly from 76 ± 42 to 45 ± 37 (P = 0.0006) by intracoronary NTP administration. IMR improved to the normal range (IMR < 30) in 9 patients (50%). Higher TIMI flow grade and lower IMR at baseline were observed more frequently in patients whose IMR recovered to normal range after NTP ad- ministration. NTP improved the microcirculatory dysfunction at the acute phase in patients who underwent PCI for STEMI and -

ALLHAT Protocol, Can Enter the Trial at the Discretion of the Principal Investigator Or His/Her Designee
Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) Protocol Revised: March 1995 May 1995 April 1998 April 2000 April 2000 Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) Protocol Table of Contents Page I. Overview............................................................................................................................ 2 II. Background........................................................................................................................ 4 III. Hypotheses and Study Power ........................................................................................... 10 IV. Eligibility and Exclusions................................................................................................. 13 V. Recruitment....................................................................................................................... 17 VI. Antihypertensive Intervention .......................................................................................... 22 VII. Cholesterol-Lowering Intervention................................................................................... 26 VIII. Laboratory Measurements ................................................................................................ 28 IX. Outcome Measurements.................................................................................................... 30 X. Study Organization .......................................................................................................... -

Multi-Omic Understanding of the Evolution of Xenobiotic Tolerance in Bacterial Isolates and Communities
Washington University in St. Louis Washington University Open Scholarship Arts & Sciences Electronic Theses and Dissertations Arts & Sciences Summer 8-15-2019 Multi-omic Understanding of the Evolution of Xenobiotic Tolerance in Bacterial Isolates and Communities Tayte Paul Campbell Washington University in St. Louis Follow this and additional works at: https://openscholarship.wustl.edu/art_sci_etds Part of the Bioinformatics Commons, Biology Commons, and the Microbiology Commons Recommended Citation Campbell, Tayte Paul, "Multi-omic Understanding of the Evolution of Xenobiotic Tolerance in Bacterial Isolates and Communities" (2019). Arts & Sciences Electronic Theses and Dissertations. 1888. https://openscholarship.wustl.edu/art_sci_etds/1888 This Dissertation is brought to you for free and open access by the Arts & Sciences at Washington University Open Scholarship. It has been accepted for inclusion in Arts & Sciences Electronic Theses and Dissertations by an authorized administrator of Washington University Open Scholarship. For more information, please contact [email protected]. WASHINGTON UNIVERSITY IN ST. LOUIS Division of Biology and Biomedical Sciences Plant and Microbial Biosciences Dissertation Examination Committee: Gautam Dantas, Chair Arpita Bose Andrew Kau Audrey Odom-John Himadri Pakrasi Fuzhong Zhang Multi-omic Understanding of the Evolution of Xenobiotic Tolerance in Bacterial Isolates and Communities by Tayte P. Campbell A dissertation presented to The Graduate School of Washington University in partial fulfillment -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

Guanfacine Extended Release for ADHD
Out of the Pipeline p Guanfacine extended release for ADHD Floyd R. Sallee, MD, PhD uanfacine extended release (GXR)— Table 1 α Once-daily a selective -2 adrenergic agonist Guanfacine extended release: GFDA-approved for the treatment formulation may of attention-defi cit/hyperactivity disor- Fast facts improve adherence der (ADHD)—has demonstrated effi cacy Brand name: Intuniv and control for inattentive and hyperactive/impulsive Indication: Attention-defi cit/hyperactivity symptoms across disorder symptom domains in 2 large trials lasting® Dowden Health Media a full day 8 and 9 weeks.1,2 GXR’s once-daily formu- Approval date: September 3, 2009 lation may increase adherence and deliver Availability date: November 2009 consistent control of symptomsCopyright across a For personalManufacturer: use Shire only full day (Table 1). Dosing forms: 1-mg, 2-mg, 3-mg, and 4-mg extended-release tablets Recommended dosage: 0.05 to 0.12 mg/kg Clinical implications once daily GXR exhibits enhancement of noradren- ergic pathways through selective direct receptor action in the prefrontal cortex.3 brain believed to play a major role in at- This mechanism of action is different from tentional and organizational functions that that of other FDA-approved ADHD medi- preclinical research has linked to ADHD.3 cations. GXR can be used alone or in com- The postsynaptic α-2A receptor is bination with stimulants or atomoxetine thought to play a central role in the opti- for treating complex ADHD, such as cases mal functioning of the PFC as illustrated accompanied by oppositional features and by the “inverted U hypothesis of PFC ac- emotional dysregulation or characterized tivation.”4 In this model, cyclic adenos- by partial stimulant response. -

Polychlorinated Biphenyls in Pigments
doi: 10.1111/cote.12167 Polychlorinated biphenyls in pigments: inadvertent production and environmental Coloration significance Technology Lisa Rodenburg,a Jia Guoa and Robert Christieb,* aDepartment of Environmental Science, Rutgers, The State University of New Jersey, New Brunswick, NJ, 08901, USA bSchool of Textiles and Design, Heriot-Watt University, Scottish Borders Campus, Netherdale, Galashiels, TD1 3HF, UK Feature article Email: [email protected] Society of Dyers and Colourists Received: 18 December 2014; Accepted: 26 June 2015 Polychlorobiphenyls are toxic, bioaccumulative, and persistent chemicals whose intentional manufacture has been banned throughout the developed world. Polychlorobiphenyls may be generated inadvertently during the production of certain pigments, including diarylides. This inadvertent production is allowed under various regulatory schemes, such as the Toxic Substances Control Act in the United States and the Stockholm Convention on Persistent Organic Pollutants. Generally, these regulations require polychlorobiphenyl levels in batches of pigment to be less than certain regulatory limits, usually 50 ppm. A growing body of evidence suggests that the use of pigments is dispersing polychlorobiphenyls throughout the environment. Polychlorobiphenyl congeners associated with pigments have been found throughout the United States in sediments and in surface waters at levels exceeding the prevailing water quality standards. A recent Japanese government study reported measured polychlorobiphenyl concentrations well