ALLHAT Protocol, Can Enter the Trial at the Discretion of the Principal Investigator Or His/Her Designee
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
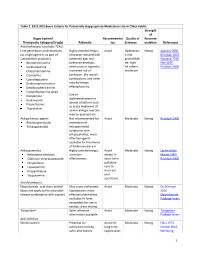
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Benign Prostatic Hyperplasia (BPH) Treatments Review 10/05/2009
Benign Prostatic Hyperplasia (BPH) Treatments Review 10/05/2009 Copyright © 2004 - 2009 by Provider Synergies, L.L.C. All rights reserved. Printed in the United States of America. All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, digital scanning, or via any information storage and retrieval system without the express written consent of Provider Synergies, L.L.C. All requests for permission should be mailed to: Attention: Copyright Administrator Intellectual Property Department Provider Synergies, L.L.C. 5181 Natorp Blvd., Suite 205 Mason, Ohio 45040 The materials contained herein represent the opinions of the collective authors and editors and should not be construed to be the official representation of any professional organization or group, any state Pharmacy and Therapeutics committee, any state Medicaid Agency, or any other clinical committee. This material is not intended to be relied upon as medical advice for specific medical cases and nothing contained herein should be relied upon by any patient, medical professional or layperson seeking information about a specific course of treatment for a specific medical condition. All readers of this material are responsible for independently obtaining medical advice and guidance from their own physician and/or other medical professional in regard to the best course of treatment for their specific medical condition. This publication, inclusive of all forms contained herein, -

Antiparasitic Properties of Cardiovascular Agents Against Human Intravascular Parasite Schistosoma Mansoni
pharmaceuticals Article Antiparasitic Properties of Cardiovascular Agents against Human Intravascular Parasite Schistosoma mansoni Raquel Porto 1, Ana C. Mengarda 1, Rayssa A. Cajas 1, Maria C. Salvadori 2 , Fernanda S. Teixeira 2 , Daniel D. R. Arcanjo 3 , Abolghasem Siyadatpanah 4, Maria de Lourdes Pereira 5 , Polrat Wilairatana 6,* and Josué de Moraes 1,* 1 Research Center for Neglected Diseases, Guarulhos University, Praça Tereza Cristina 229, São Paulo 07023-070, SP, Brazil; [email protected] (R.P.); [email protected] (A.C.M.); [email protected] (R.A.C.) 2 Institute of Physics, University of São Paulo, São Paulo 05508-060, SP, Brazil; [email protected] (M.C.S.); [email protected] (F.S.T.) 3 Department of Biophysics and Physiology, Federal University of Piaui, Teresina 64049-550, PI, Brazil; [email protected] 4 Ferdows School of Paramedical and Health, Birjand University of Medical Sciences, Birjand 9717853577, Iran; [email protected] 5 CICECO-Aveiro Institute of Materials & Department of Medical Sciences, University of Aveiro, 3810-193 Aveiro, Portugal; [email protected] 6 Department of Clinical Tropical Medicine, Faculty of Tropical Medicine, Mahidol University, Bangkok 10400, Thailand * Correspondence: [email protected] (P.W.); [email protected] (J.d.M.) Citation: Porto, R.; Mengarda, A.C.; Abstract: The intravascular parasitic worm Schistosoma mansoni is a causative agent of schistosomiasis, Cajas, R.A.; Salvadori, M.C.; Teixeira, a disease of great global public health significance. Praziquantel is the only drug available to F.S.; Arcanjo, D.D.R.; Siyadatpanah, treat schistosomiasis and there is an urgent demand for new anthelmintic agents. -

Guideline for Preoperative Medication Management
Guideline: Preoperative Medication Management Guideline for Preoperative Medication Management Purpose of Guideline: To provide guidance to physicians, advanced practice providers (APPs), pharmacists, and nurses regarding medication management in the preoperative setting. Background: Appropriate perioperative medication management is essential to ensure positive surgical outcomes and prevent medication misadventures.1 Results from a prospective analysis of 1,025 patients admitted to a general surgical unit concluded that patients on at least one medication for a chronic disease are 2.7 times more likely to experience surgical complications compared with those not taking any medications. As the aging population requires more medication use and the availability of various nonprescription medications continues to increase, so does the risk of polypharmacy and the need for perioperative medication guidance.2 There are no well-designed trials to support evidence-based recommendations for perioperative medication management; however, general principles and best practice approaches are available. General considerations for perioperative medication management include a thorough medication history, understanding of the medication pharmacokinetics and potential for withdrawal symptoms, understanding the risks associated with the surgical procedure and the risks of medication discontinuation based on the intended indication. Clinical judgement must be exercised, especially if medication pharmacokinetics are not predictable or there are significant risks associated with inappropriate medication withdrawal (eg, tolerance) or continuation (eg, postsurgical infection).2 Clinical Assessment: Prior to instructing the patient on preoperative medication management, completion of a thorough medication history is recommended – including all information on prescription medications, over-the-counter medications, “as needed” medications, vitamins, supplements, and herbal medications. Allergies should also be verified and documented. -

Comparison of Different Alpha-Blocker Combinations in Male Hypertensives with Refractory Lower Urinary Tract Symptoms
대한남성과학회지:제 29 권 제 3 호 2011년 12월 Korean J Androl. Vol. 29, No. 3, December 2011 http://dx.doi.org/10.5534/kja.2011.29.3.242 Comparison of Different Alpha-blocker Combinations in Male Hypertensives with Refractory Lower Urinary Tract Symptoms Keon Cheol Lee1, Jong Gu Kim2, Sung Yong Cho1, Joon Sung Jeon1, In Rae Cho1 Department of Urology, 1Inje University Ilsanpaik Hospital, Goyang, 2Happy Urology Clinic, Ansan, Korea =Abstract= Purpose: We compared the efficacy and safety profiles of dose increase, traditional combination methods, and combining different alpha blockers in hypertensive males with lower urinary tract symptom (LUTS) refractory to an initial dose of 4 mg doxazosin. Materials and Methods: Between 2000 and 2005, 374 male patients with LUTS and hypertension unresponsive to 4 weeks of 4 mg doxazosin were enrolled. The subjects were randomly classified into 3 groups, 8 mg/day of doxazosin (D group), 4 mg of doxazosin plus 0.2 mg/day of tamsulosin (DT group), and 4 mg doxazosin plus 5 mg/day finasteride (DF group). Patients were evaluated based on their International Prostate Symptom Score (IPSS), quality of life (QOL), uroflowmetry and blood pressure (BP) and adverse events (AEs) at the baseline and 3 and 12 months after treatment. Results: The 269 patients (71.9%) were followed for at least 1 year (D group n=84, DT group n=115, and DF group n=70). The clinical parameters before and after initial 4 mg/day doxazosin were not different among the 3 groups. IPSS improvement after 3 months and maximal flow rate (Qmax) improvement after 3 and 12 months were significantly higher in the D and DT groups than the DF group (p<0.05). -

Different Effects of Propranolol, Bisoprolol, Carvedilol and Doxazosin on Heart Rate, Blood Pressure, and Plasma Concentrations of Epinephrine and Norepinephrine K
Journal of Clinical and Basic Cardiology An Independent International Scientific Journal Journal of Clinical and Basic Cardiology 2003; 6 (1-4), 69-72 Different Effects of Propranolol Bisoprolol, Carvedilol and Doxazosin on Heart Rate, Blood Pressure, and Plasma Concentrations of Epinephrine and Norepinephrine Stoschitzky K, Donnerer J, Klein W, Koshucharova G Kraxner W, Lercher P, Maier R, Watzinger N, Zweiker R Homepage: www.kup.at/jcbc Online Data Base Search for Authors and Keywords Indexed in Chemical Abstracts EMBASE/Excerpta Medica Krause & Pachernegg GmbH · VERLAG für MEDIZIN und WIRTSCHAFT · A-3003 Gablitz/Austria ORIGINAL PAPERS, CLINICAL CARDIOLOGY Alpha- Versus Beta-Blockers J Clin Basic Cardiol 2003; 6: 69 Different Effects of Propranolol, Bisoprolol, Carvedilol and Doxazosin on Heart Rate, Blood Pressure, and Plasma Concentrations of Epinephrine and Norepinephrine K. Stoschitzky1, G. Koshucharova1, R. Zweiker1, P. Lercher1, R. Maier1, N. Watzinger1, W. Kraxner1, W. Klein1, J. Donnerer2 Background: Despite of its beta-blocking effects, carvedilol has been shown not to decrease resting heart rate in healthy subjects. Therefore, we compared haemodynamic effects of carvedilol (an alpha- and beta-blocker), propranolol (a non-selec- tive beta-blocker), bisoprolol (a beta1-selective beta-blocker), doxazosin (an alpha-blocker) and placebo, at rest and during exercise. In addition, we measured plasma levels of epinephrine and norepinephrine. Methods: Twelve healthy males received single oral doses of 80 mg propranolol, 5 mg bisoprolol, 50 mg carvedilol, 4 mg doxazosin and placebo according to a randomized, double-blind, crossover protocol. Three hours after drug intake, heart rate and blood pressure were measured at rest, after 10 min of exercise, and after 15 min of recovery. -

Initial Medication Selection for Treatment of Hypertension in an Open-Panel HMO
J Am Board Fam Pract: first published as 10.3122/jabfm.8.1.1 on 1 January 1995. Downloaded from Initial Medication Selection For Treatment Of Hypertension In An Open-Panel HMO Micky jerome, PharmD, MBA, George C. Xakellis, MD, Greg Angstman, MD, and Wayne Patchin, MBA Background: During the past 25 years recommendations for treating hypertension have evolved from a stepped-care approach to monotherapy or sequential monotherapy as experience has been gained and new antihypertensive agents have been introduced. In an effort to develop a disease management strategy for hypertension, we investigated the prescribing patterns of initial medication therapy for newly treated hypertensive patients. Methods: We examined paid claims data of an open-panel HMO located in the midwest. Charts from 377 patients with newly treated hypertension from a group of 12,242 hypertenSive patients in a health insurance population of 85,066 persons were studied. The type of medication regimen received by patients newly treated for hypertension during an 18-month period was categorized into monotherapy, sequential monotherapy, stepped care, and initial treatment with multiple agents. With monotherapy, the class of medication was also reported. Associations between use of angiotensin-converting enzyme (ACE) inhibitors, calcium channel blockers, or (3-blockers and presence of comorbid conditions were reported. Results: Fifty-five percent of patients received monotherapy, 22 percent received stepped care, and 18 percent received sequential monotherapy. Of those 208 patients receiving monotherapy, 30 percent were prescribed a calcium channel blocker, 22 percent an ACE inhibitor, and 14 percent a f3-blocker. No customization of treatment for comorbid conditions was noted. -

Preventive Drug List
PREVENTIVE DRUG LIST Preventive medications are used for the prevention of conditions such as high blood pressure, high cholesterol, diabetes, asthma, osteoporosis, heart attack, stroke and prenatal nutrient deficiency. Following is a list of generic preventive medications covered at 100%, arranged by type of condition. diltiazem sotalol AF glipizide Asthma related diltiazem ER sotalol HCl glipizide ER albuterol sulfate doxazosin mesylate spironolactone glipizide/metformin HCl albuterol sulfate (nebulizer enalapril maleate spironolactone/hctz glyburide solution) enalapril maleate/hctz telmisartan glyburide micronized albuterol sulfate/ipratropium eplerenone telmisartan/amlodipine glyburide/metformin nebulizer solution eprosartan mesylate telmisartan/hctz Humalog budesonide felodipine ER terazosin HCl Humulin caffeine citrate fosinopril sodium timolol maleate Invokamet cromolyn sodium inhalation fosinopril sodium/hctz torsemide Invokamet XR solution furosemide trandolapril Invokana ipratropium bromide guanfacine HCl trandolapril/verapamil Janumet levalbuterol HCl hydralazine HCl triamterene/hctz Janumet XR levalbuterol tartrate HFA hydrochlorothiazide valsartan Januvia metaproterenol sulfate indapamide valsartan/hctz Kombiglyze XR montelukast irbesartan Vecamyl – mecamylamine HCl Lantus terbutaline sulfate irbesartan/hctz Verapamil Lantus SoloStar TheoChron isradipine Levemir theophylline anhydrous labetalol HCl Blood thinner related Levemir FlexTouch zafirlukast lisinopril aspirin/dipyridamole ER metformin HCl lisinopril/hctz cilostazol -

Information for the User Doxazosin STADA 1 Mg, 2 Mg, 4 Mg Tablets
DE/H/0287/001-003 Package leaflet: Information for the user Doxazosin STADA 1 mg, 2 mg, 4 mg tablets Doxazosin Read all of this leaflet carefully before you start taking this medicine because it contains important information for you. Keep this leaflet. You may need to read it again. If you have any further questions, ask your doctor or pharmacist. This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours. If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4. In this leaflet 1. What Doxazosin STADA is and what it is used for 2. What you need to know before you take Doxazosin STADA 3. How to take Doxazosin STADA 4. Possible side effects 5. How to store Doxazosin STADA 6. Contents of the pack and other information 1. What Doxazosin STADA is and what it is used for The active ingredient in your tablets, doxazosin, belongs to a group of medicines known as alpha-1 antagonists. Doxazosin STADA is used to treat the following conditions. High blood pressure (essential hypertension). If left uncontrolled, high blood pressure can increase the risk of heart disease or stroke. Doxazosin STADA works by widening your blood vessels making it easier for your heart to pump blood through them. This helps to lower raised blood pressure and reduce the risk of heart disease. Symptoms of benign prostatic hyperplasia (BPH). -

Provider- Pain Quick Reference Guide
A QUICK REFERENCE GUIDE (2019) PBM Academic Detailing Service Posttraumatic Stress Disorder A VA Clinician's Guide to Optimal Treatment of Posttraumatic Stress Disorder (PTSD) VA PBM Academic Detailing Service Real Provider Resources Real Patient Results Your Partner in Enhancing Veteran Health Outcomes VA PBM Academic Detailing Service Email Group [email protected] VA PBM Academic Detailing Service SharePoint Site https://vaww.portal2.va.gov/sites/ad VA PBM Academic Detailing Public Website http://www.pbm.va.gov/PBM/academicdetailingservicehome.asp Table of Contents Abbreviations . 1 PTSD Treatment Decision Aid . 3 VA/DoD 2017 Clinical Practice Guideline: Treatment of PTSD . 4 First-Line Treatment: Trauma-focused Psychotherapies with the Strongest Evidence . 5 First-Line Treatment: Trauma-Focused Psychotherapies with Sufficient Evidence . 6 Comparison of Antidepressants Studied in PTSD . 7 Recommended Antidepressant For PTSD: Dosing . 9 Additional Medications Studied in PTSD: Dosing . 11 Switching Antidepressants . 12 Antidepressants and Sexual Dysfunction . 14 i TOC (continued) Sexual Dysfunction Treatment Strategies . 16 Antidepressants and Hyponatremia . 17 Additional Pharmacotherapy Options for Veterans Refractory to Standard Treatments . 19 Discussing Benzodiazepine Withdrawal . 21 Prazosin Tips . 25 Prazosin Precautions . 26 Managing PTSD Nightmares in Veterans with LUTS Associated with BPH . 27 Other Medications Studied in PTSD-Related Nightmares . 29 References . 30 ii Abbreviations Anti-Ach = anticholinergic -

Prazosin and Doxazosin for PTSD Are Underutilized and Underdosed
Guest Editorial Prazosin and doxazosin for PTSD are underutilized and underdosed Maju Mathew Koola, MD Often, clinicians tend to focus on the • 10% among Gulf War veterans diagnosis and treatment of psychi- • 14% among Iraq and Afghanistan veterans.3 atric disorders, such as psychosis, bipolar disorder, depression, anxiety, Why is PTSD overlooked insomnia, and substance use disor- in substance use? ders (SUD), but posttraumatic stress Among individuals with SUD, 10% to 4 disorder (PTSD) often is overlooked 63% have comorbid PTSD. A recent report underscores the complexity and and underdiagnosed,1 especially Prazosin and doxazosin, challenges of SUD–PTSD comorbidity.5 when comorbid with another psy- Most PTSD patients with comorbid which have been used chiatric disorder such as SUD. SUD receive treatment only for SUD for PTSD, also can treat and the PTSD symptoms often are The primary symptoms of PTSD are unaddressed.5 Those suffering from substance abuse; this recurrent and include intrusive memo- PTSD often abuse alcohol because they ‘two birds with one stone’ ries and dreams of the traumatic events, might consider it to be a coping strat- approach could become flashbacks, hypervigilance, irritabil- egy. Alcohol reduces hyperactivation ity, sleep disturbances, and persistent of the dorsal anterior cingulate cor- more common in clinical avoidance of stimuli associated with tex caused by re-experiencing PTSD practice the traumatic event. According to the symptoms. Other substances of abuse, National Comorbidity Survey, the esti- such as Cannabis, could suppress PTSD mated lifetime prevalence of PTSD symptoms through alternate mecha- among adults is 6.8% and is more com- nisms (eg, endocannabinoid recep- mon in women (9.7%) than men (3.6%).2 tors). -

Benign Prostatic Hyperplasia
Benign Prostatic Hyperplasia (BPH) Treatments Therapeutic Class Review (TCR) November 18, 2019 No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, digital scanning, or via any information storage or retrieval system without the express written consent of Magellan Rx Management. All requests for permission should be mailed to: Magellan Rx Management Attention: Legal Department 6950 Columbia Gateway Drive Columbia, Maryland 21046 The materials contained herein represent the opinions of the collective authors and editors and should not be construed to be the official representation of any professional organization or group, any state Pharmacy and Therapeutics committee, any state Medicaid Agency, or any other clinical committee. This material is not intended to be relied upon as medical advice for specific medical cases and nothing contained herein should be relied upon by any patient, medical professional or layperson seeking information about a specific course of treatment for a specific medical condition. All readers of this material are responsible for independently obtaining medical advice and guidance from their own physician and/or other medical professional in regard to the best course of treatment for their specific medical condition. This publication, inclusive of all forms contained herein, is intended to be educational in nature and is intended to be used for informational purposes only. Send comments and suggestions to [email protected].