Adiabatic Thermodynamics of Fluids
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
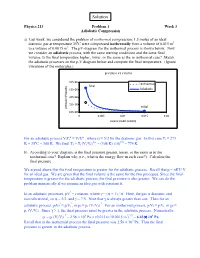
3-1 Adiabatic Compression
Solution Physics 213 Problem 1 Week 3 Adiabatic Compression a) Last week, we considered the problem of isothermal compression: 1.5 moles of an ideal diatomic gas at temperature 35oC were compressed isothermally from a volume of 0.015 m3 to a volume of 0.0015 m3. The pV-diagram for the isothermal process is shown below. Now we consider an adiabatic process, with the same starting conditions and the same final volume. Is the final temperature higher, lower, or the same as the in isothermal case? Sketch the adiabatic processes on the p-V diagram below and compute the final temperature. (Ignore vibrations of the molecules.) α α For an adiabatic process ViTi = VfTf , where α = 5/2 for the diatomic gas. In this case Ti = 273 o 1/α 2/5 K + 35 C = 308 K. We find Tf = Ti (Vi/Vf) = (308 K) (10) = 774 K. b) According to your diagram, is the final pressure greater, lesser, or the same as in the isothermal case? Explain why (i.e., what is the energy flow in each case?). Calculate the final pressure. We argued above that the final temperature is greater for the adiabatic process. Recall that p = nRT/ V for an ideal gas. We are given that the final volume is the same for the two processes. Since the final temperature is greater for the adiabatic process, the final pressure is also greater. We can do the problem numerically if we assume an idea gas with constant α. γ In an adiabatic processes, pV = constant, where γ = (α + 1) / α. -

Thermodynamics Formulation of Economics Burin Gumjudpai A
Thermodynamics Formulation of Economics 306706052 NIDA E-THESIS 5710313001 thesis / recv: 09012563 15:07:41 seq: 16 Burin Gumjudpai A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Economics (Financial Economics) School of Development Economics National Institute of Development Administration 2019 Thermodynamics Formulation of Economics Burin Gumjudpai School of Development Economics Major Advisor (Associate Professor Yuthana Sethapramote, Ph.D.) The Examining Committee Approved This Thesis Submitted in Partial 306706052 Fulfillment of the Requirements for the Degree of Master of Economics (Financial Economics). Committee Chairperson NIDA E-THESIS 5710313001 thesis / recv: 09012563 15:07:41 seq: 16 (Assistant Professor Pongsak Luangaram, Ph.D.) Committee (Assistant Professor Athakrit Thepmongkol, Ph.D.) Committee (Associate Professor Yuthana Sethapramote, Ph.D.) Dean (Associate Professor Amornrat Apinunmahakul, Ph.D.) ______/______/______ ABST RACT ABSTRACT Title of Thesis Thermodynamics Formulation of Economics Author Burin Gumjudpai Degree Master of Economics (Financial Economics) Year 2019 306706052 We consider a group of information-symmetric consumers with one type of commodity in an efficient market. The commodity is fixed asset and is non-disposable. We are interested in seeing if there is some connection of thermodynamics formulation NIDA E-THESIS 5710313001 thesis / recv: 09012563 15:07:41 seq: 16 to microeconomics. We follow Carathéodory approach which requires empirical existence of equation of state (EoS) and coordinates before performing maximization of variables. In investigating EoS of various system for constructing the economics EoS, unexpectedly new insights of thermodynamics are discovered. With definition of truly endogenous function, criteria rules and diagrams are proposed in identifying the status of EoS for an empirical equation. -

The First Law of Thermodynamics Continued Pre-Reading: §19.5 Where We Are
Lecture 7 The first law of thermodynamics continued Pre-reading: §19.5 Where we are The pressure p, volume V, and temperature T are related by an equation of state. For an ideal gas, pV = nRT = NkT For an ideal gas, the temperature T is is a direct measure of the average kinetic energy of its 3 3 molecules: KE = nRT = NkT tr 2 2 2 3kT 3RT and vrms = (v )av = = r m r M p Where we are We define the internal energy of a system: UKEPE=+∑∑ interaction Random chaotic between atoms motion & molecules For an ideal gas, f UNkT= 2 i.e. the internal energy depends only on its temperature Where we are By considering adding heat to a fixed volume of an ideal gas, we showed f f Q = Nk∆T = nR∆T 2 2 and so, from the definition of heat capacity Q = nC∆T f we have that C = R for any ideal gas. V 2 Change in internal energy: ∆U = nCV ∆T Heat capacity of an ideal gas Now consider adding heat to an ideal gas at constant pressure. By definition, Q = nCp∆T and W = p∆V = nR∆T So from ∆U = Q W − we get nCV ∆T = nCp∆T nR∆T − or Cp = CV + R It takes greater heat input to raise the temperature of a gas a given amount at constant pressure than constant volume YF §19.4 Ratio of heat capacities Look at the ratio of these heat capacities: we have f C = R V 2 and f + 2 C = C + R = R p V 2 so C p γ = > 1 CV 3 For a monatomic gas, CV = R 3 5 2 so Cp = R + R = R 2 2 C 5 R 5 and γ = p = 2 = =1.67 C 3 R 3 YF §19.4 V 2 Problem An ideal gas is enclosed in a cylinder which has a movable piston. -

Adiabatic Bulk Moduli
8.03 at ESG Supplemental Notes Adiabatic Bulk Moduli To find the speed of sound in a gas, or any property of a gas involving elasticity (see the discussion of the Helmholtz oscillator, B&B page 22, or B&B problem 1.6, or French Pages 57-59.), we need the “bulk modulus” of the fluid. This will correspond to the “spring constant” of a spring, and will give the magnitude of the restoring agency (pressure for a gas, force for a spring) in terms of the change in physical dimension (volume for a gas, length for a spring). It turns out to be more useful to use an intensive quantity for the bulk modulus of a gas, so what we want is the change in pressure per fractional change in volume, so the bulk modulus, denoted as κ (the Greek “kappa” which, when written, has a great tendency to look like k, and in fact French uses “K”), is ∆p dp κ = − , or κ = −V . ∆V/V dV The minus sign indicates that for normal fluids (not all are!), a negative change in volume results in an increase in pressure. To find the bulk modulus, we need to know something about the gas and how it behaves. For our purposes, we will need three basic principles (actually 2 1/2) which we get from thermodynamics, or the kinetic theory of gasses. You might well have encountered these in previous classes, such as chemistry. A) The ideal gas law; pV = nRT , with the standard terminology. B) The first law of thermodynamics; dU = dQ − pdV,whereUis internal energy and Q is heat. -

Session 15 Thermodynamics
Session 15 Thermodynamics Cheryl Hurkett Physics Innovations Centre for Excellence in Learning and Teaching CETL (Leicester) Department of Physics and Astronomy University of Leicester 1 Contents Welcome .................................................................................................................................. 4 Session Authors .................................................................................................................. 4 Learning Objectives .............................................................................................................. 5 The Problem ........................................................................................................................... 6 Issues .................................................................................................................................... 6 Energy, Heat and Work ........................................................................................................ 7 The First Law of thermodynamics................................................................................... 8 Work..................................................................................................................................... 9 Heat .................................................................................................................................... 11 Principal Specific heats of a gas ..................................................................................... 12 Summary .......................................................................................................................... -

1 Microphysics
ASTR 501 Stellar Physics 1 1 Microphysics • equation of state, density: ρ(P, T, X) • radiative absorption coefficient, opacity: κ(P, T, X) • rate of energy production: (P, T, X) (nuclear physics) Nuclear physics also responsible for composition changes dXj = Fˆj · Xj (1) dt burn while the total composition change also includes a mixing term: dX ∂ dX i = 4πr2ρ D i (2) dt mix ∂m dm 1.1 Equation of state For the fluid to have an EOS it must be collisional: lregion λ (3) where λ is the mean free path. P from equation of state, consider three cases: ASTR 501 Stellar Physics 2 (a) ideal gas equation of state: p = R ρT (b) barotropic equation of state: pressure p depends only on ρ, as for example • isothermal: p ∼ ρ • adiabatic: p = Kργ Plus non-ideal effects, e.g. electron degeneracy. 1.2 Ideal gas equation of state Assumption: internal energy entirely in kinetic energy according to kinetic theory: = CVT , where CV is the specific heat at constant volume. This can be seen from the first law of thermodynamics dQ = d + pdV , where V is the specific volume, d if we express d = dT dT . d Similarly, the specific heat at constant pressure Cp = dT + R where R is the gas constant. This follows from the ideal gas equation of state. [†13] It also follows that Cp − CV = R [†14], and the ratio of specific heats is defined as C γ = p . (4) CV ASTR 501 Stellar Physics 3 α CV can be related to R as CV = 2 R , where α is the number of degrees of freedom 1 per particle, since the energy per particle per degree of freedom is 2kT where k is 2+α the Boltzmann constant, and R = nk. -

Heat Capacity Ratio of Gases
Heat Capacity Ratio of Gases Carson Hasselbrink [email protected] Office Hours: Mon 10-11am, Beaupre 360 1 Purpose • To determine the heat capacity ratio for a monatomic and a diatomic gas. • To understand and mathematically model reversible & irreversible adiabatic processes for ideal gases. • To practice error propagation for complex functions. 2 Key Physical Concepts • Heat capacity is the amount of heat required to raise the temperature of an object or substance one degree 풅풒 푪 = 풅푻 • Heat Capacity Ratio is the ratio of specific heats at constant pressure and constant volume 퐶푝 훾 = 퐶푣 휕퐻 휕퐸 Where 퐶 = ( ) and 퐶 = ( ) 푝 휕푇 푝 푣 휕푇 푣 • An adiabatic process occurs when no heat is exchanged between the system and the surroundings 3 Theory: Heat Capacity 휕퐸 • Heat Capacity (Const. Volume): 퐶 = ( ) /푛 푣,푚 휕푇 푣 3푅푇 3푅 – Monatomic: 퐸 = , so 퐶 = 푚표푛푎푡표푚푖푐 2 푣,푚 2 5푅푇 5푅 – Diatomic: 퐸 = , so 퐶 = 푑푖푎푡표푚푖푐 2 푣,푚 2 • Heat Capacity Ratio: 퐶푝,푚 = 퐶푣,푚 + 푅 퐶푝,푚 푅 훾 = = 1 + 퐶푣,푚 퐶푣,푚 푃 [ln 1 ] 푃2 – Reversible: 훾 = 푃 [ln 1 ] 푃3 푃 [ 1 −1] 푃2 – Irreversible: 훾 = 푃 [ 1 −1] 푃3 • Diatomic heat capacity > Monatomic Heat Capacity 4 Theory: Determination of Heat Capacity Ratio • We will subject a gas to an adiabatic expansion and then allow the gas to return to its original temperature via an isochoric process, during which time it will cool. • This expansion and warming can be modeled in two different ways. 5 Reversible Expansion (Textbook) • Assume that pressure in carboy (P1) and exterior pressure (P2) are always close enough that entire process is always in equilibrium • Since system is in equilibrium, each step must be reversible 6 Irreversible Expansion (Lab Syllabus) • Assume that pressure in carboy (P1) and exterior pressure (P2) are not close enough; there is sudden deviation in pressure; the system is not in equilibrium • Since system is not in equilibrium, the process becomes irreversible. -

12/8 and 12/10/2010
PY105 C1 1. Help for Final exam has been posted on WebAssign. 2. The Final exam will be on Wednesday December 15th from 6-8 pm. First Law of Thermodynamics 3. You will take the exam in multiple rooms, divided as follows: SCI 107: Abbasi to Fasullo, as well as Khajah PHO 203: Flynn to Okuda, except for Khajah SCI B58: Ordonez to Zhang 1 2 Heat and Work done by a Gas Thermodynamics Initial: Consider a cylinder of ideal Thermodynamics is the study of systems involving gas at room temperature. Suppose the piston on top of energy in the form of heat and work. the cylinder is free to move vertically without friction. When the cylinder is placed in a container of hot water, heat Equilibrium: is transferred into the cylinder. Where does the heat energy go? Why does the volume increase? 3 4 The First Law of Thermodynamics The First Law of Thermodynamics The First Law is often written as: Some of the heat energy goes into raising the temperature of the gas (which is equivalent to raising the internal energy of the gas). The rest of it does work by raising the piston. ΔEQWint =− Conservation of energy leads to: QEW=Δ + int (the first law of thermodynamics) This form of the First Law says that the change in internal energy of a system is Q is the heat added to a system (or removed if it is negative) equal to the heat supplied to the system Eint is the internal energy of the system (the energy minus the work done by the system (usually associated with the motion of the atoms and/or molecules). -

Astronomy 112: the Physics of Stars Class 6 Notes: Internal Energy And
Astronomy 112: The Physics of Stars Class 6 Notes: Internal Energy and Radiative Transfer In the last class we used the kinetic theory of gasses to understand the pressure of stellar material. The kinetic view is essential to generalizing the concept of pressure to the environ- ments found in stars, where gas can be relativistic, degenerate, or both. The goal of today’s class is to extend that kinetic picture by thinking about pressure in stars in terms of the associated energy content of the gas. This will let us understand how the energy flow in stars interacts with gas pressure, a crucial step toward building stellar models. I. Pressure and Energy A. The Relationship Between Pressure and Internal Energy The fundamental object we dealt with in the last class was the distribution of particle mometa, dn(p)/dp, which we calculated from the Boltzmann distribu- tion. Given this, we could compute the pressure. However, this distribution also corresponds to a specific energy content, since for particles that don’t have inter- nal energy states, internal energy is just the kinetic energy of particle motions. Given a distribution of particle momenta dn(p)/dp in some volume of space, the corresponding density of energy within that volume of space is Z ∞ dn(p) e = (p) dp, 0 dp where (p) is the energy of a particle with momentum p. It is often more conve- nient to think about the energy per unit mass than the energy per unit volume. The energy per unit mass is just e divided by the density: 1 Z ∞ dn(p) u = (p) dp. -

2Nd Meeting on Energy and Environmental Economics 18Th September 2015 DEGEI, Universidade De Aveiro
2nd Meeting on Energy and Environmental Economics 18th September 2015 DEGEI, Universidade de Aveiro REVISITING COMPRESSED AIR ENERGY STORAGE Catarina Matos1, Patrícia Pereira da Silva2, Júlio Carneiro3 1 MIT-Portugal Program, University of Coimbra and ICT- Instituto de Ciências da Terra, University of Évora (Portugal); [email protected] 2Faculty of Economics, University of Coimbra and INESC, Coimbra (Portugal); [email protected] 3 Departamento de Geociências, Escola de Ciências e Formação Avançada, Instituto de Investigação e Formação Avançada, Instituto de Ciências da Terra, Universidade de Évora - Portugal; [email protected] ABSTRACT The use of renewable energies as a response to the EU targets defined for 2030 Climate Change and Energy has been increasing. Also non-dispatchable and intermittent renewable energies like wind and solar cannot generally match supply and demand, which can also cause some problems in the grid. So, the increased interest in energy storage has evolved and there is nowadays an urgent need for larger energy storage capacity. Compressed Air Energy Storage (CAES) is a proven technology for storing large quantities of electrical energy in the form of high-pressure air for later use when electricity is needed. It exists since the 1970’s and is one of the few energy storage technologies suitable for long duration (tens of hours) and utility scale (hundreds to thousands of MW) applications. It is also one of the most cost-effective solutions for large to small scale storage applications. Compressed Air Energy Storage can be integrated and bring advantages to different levels of the electric system, from the Generation level, to the Transmission and Distribution levels, so in this paper a revisit of CAES is done in order to better understand what and how it can be used for our modern needs of energy storage. -

Development and Formative Evaluation of an Instructional Simulation of Adiabatic Processes Ying-Shao Hsu Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1997 Development and formative evaluation of an instructional simulation of adiabatic processes Ying-Shao Hsu Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Curriculum and Instruction Commons, Educational Psychology Commons, Higher Education and Teaching Commons, and the Science and Mathematics Education Commons Recommended Citation Hsu, Ying-Shao, "Development and formative evaluation of an instructional simulation of adiabatic processes " (1997). Retrospective Theses and Dissertations. 11992. https://lib.dr.iastate.edu/rtd/11992 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfihn master. UMl fibns the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter &ce, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. -

«Modeling of the Adiabatic and Isothermal Methanation Process»
Scientific Journal of Riga Technical University Environmental and Climate Technologies DOI: 10.2478/v10145-011-0011-5 2011 _________________________________________________________________________________________________ Volume 6 Modeling of the Adiabatic and Isothermal Methanation Process Jekaterina Porubova1, Gatis Bazbauers2, Darja Markova3, 1-3Riga Technical University, Institute of Energy Systems and Environment Abstract – Increased use of biomass offers one of the ways to reduce anthropogenic impact on the environment. Using The main differences between these two methods are that, various biomass conversion processes, it is possible to obtain during the thermo-chemical conversion, biomass gasification different types of fuels: is followed by the methanation process, but bio-gas • solid, e.g. bio-carbon; production occurs during the anaerobic digestion of biomass. • liquid, e.g. biodiesel and ethanol; In addition, thermo-chemical transformation is characterized • gaseous, e.g. biomethane. by a higher efficiency compared to bio-chemical conversion. Biomethane can be used in the transport and energy sector, This higher efficiency leads to a faster transformation and the total methane production efficiency can reach 65%. By response, which is a few seconds or minutes for the thermo- modeling adiabatic and isothermal methanation processes, the chemical conversion and several days, weeks or even more most effective one from the methane production point of view is defined. Influence of the process parameters on the overall for the anaerobic digestion of biomass [11]. efficiency of the methane production is determined. Below are the main methanation reactions [5,6]: Keywords – biomethane, methanation, Aspen Plus®, CO + 3H2 ↔ CH4 + H2O ∆HR = -206.28 kJ/mol (1.1) modeling CO2 + 4H2 ↔ CH4 + 2H2O ∆HR = -165.12 kJ/mol (1.2) I.