Prostaglandin D2 and the Role of the DP1, DP2 and TP Receptors in the Control of Airway Reflex Events
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

4 Supplementary File
Supplemental Material for High-throughput screening discovers anti-fibrotic properties of Haloperidol by hindering myofibroblast activation Michael Rehman1, Simone Vodret1, Luca Braga2, Corrado Guarnaccia3, Fulvio Celsi4, Giulia Rossetti5, Valentina Martinelli2, Tiziana Battini1, Carlin Long2, Kristina Vukusic1, Tea Kocijan1, Chiara Collesi2,6, Nadja Ring1, Natasa Skoko3, Mauro Giacca2,6, Giannino Del Sal7,8, Marco Confalonieri6, Marcello Raspa9, Alessandro Marcello10, Michael P. Myers11, Sergio Crovella3, Paolo Carloni5, Serena Zacchigna1,6 1Cardiovascular Biology, 2Molecular Medicine, 3Biotechnology Development, 10Molecular Virology, and 11Protein Networks Laboratories, International Centre for Genetic Engineering and Biotechnology (ICGEB), Padriciano, 34149, Trieste, Italy 4Institute for Maternal and Child Health, IRCCS "Burlo Garofolo", Trieste, Italy 5Computational Biomedicine Section, Institute of Advanced Simulation IAS-5 and Institute of Neuroscience and Medicine INM-9, Forschungszentrum Jülich GmbH, 52425, Jülich, Germany 6Department of Medical, Surgical and Health Sciences, University of Trieste, 34149 Trieste, Italy 7National Laboratory CIB, Area Science Park Padriciano, Trieste, 34149, Italy 8Department of Life Sciences, University of Trieste, Trieste, 34127, Italy 9Consiglio Nazionale delle Ricerche (IBCN), CNR-Campus International Development (EMMA- INFRAFRONTIER-IMPC), Rome, Italy This PDF file includes: Supplementary Methods Supplementary References Supplementary Figures with legends 1 – 18 Supplementary Tables with legends 1 – 5 Supplementary Movie legends 1, 2 Supplementary Methods Cell culture Primary murine fibroblasts were isolated from skin, lung, kidney and hearts of adult CD1, C57BL/6 or aSMA-RFP/COLL-EGFP mice (1) by mechanical and enzymatic tissue digestion. Briefly, tissue was chopped in small chunks that were digested using a mixture of enzymes (Miltenyi Biotec, 130- 098-305) for 1 hour at 37°C with mechanical dissociation followed by filtration through a 70 µm cell strainer and centrifugation. -
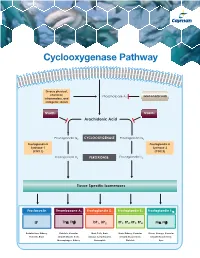
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Cr
Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Crizotinib (PF-02341066) 1 4 55 Docetaxel 1 5 98 Anastrozole 1 6 25 Cladribine 1 7 23 Methotrexate 1 8 -187 Letrozole 1 9 65 Entecavir Hydrate 1 10 48 Roxadustat (FG-4592) 1 11 19 Imatinib Mesylate (STI571) 1 12 0 Sunitinib Malate 1 13 34 Vismodegib (GDC-0449) 1 14 64 Paclitaxel 1 15 89 Aprepitant 1 16 94 Decitabine 1 17 -79 Bendamustine HCl 1 18 19 Temozolomide 1 19 -111 Nepafenac 1 20 24 Nintedanib (BIBF 1120) 1 21 -43 Lapatinib (GW-572016) Ditosylate 1 22 88 Temsirolimus (CCI-779, NSC 683864) 1 23 96 Belinostat (PXD101) 1 24 46 Capecitabine 1 25 19 Bicalutamide 1 26 83 Dutasteride 1 27 68 Epirubicin HCl 1 28 -59 Tamoxifen 1 29 30 Rufinamide 1 30 96 Afatinib (BIBW2992) 1 31 -54 Lenalidomide (CC-5013) 1 32 19 Vorinostat (SAHA, MK0683) 1 33 38 Rucaparib (AG-014699,PF-01367338) phosphate1 34 14 Lenvatinib (E7080) 1 35 80 Fulvestrant 1 36 76 Melatonin 1 37 15 Etoposide 1 38 -69 Vincristine sulfate 1 39 61 Posaconazole 1 40 97 Bortezomib (PS-341) 1 41 71 Panobinostat (LBH589) 1 42 41 Entinostat (MS-275) 1 43 26 Cabozantinib (XL184, BMS-907351) 1 44 79 Valproic acid sodium salt (Sodium valproate) 1 45 7 Raltitrexed 1 46 39 Bisoprolol fumarate 1 47 -23 Raloxifene HCl 1 48 97 Agomelatine 1 49 35 Prasugrel 1 50 -24 Bosutinib (SKI-606) 1 51 85 Nilotinib (AMN-107) 1 52 99 Enzastaurin (LY317615) 1 53 -12 Everolimus (RAD001) 1 54 94 Regorafenib (BAY 73-4506) 1 55 24 Thalidomide 1 56 40 Tivozanib (AV-951) 1 57 86 Fludarabine -

Effect of TTC-909 on Cerebral Infarction Following Permanent Occlusion of the Middle Cerebral Artery in Stroke Prone Spontaneously Hypertensive Rats
J Pharmacol Sci 91, 305 – 312 (2003) Journal of Pharmacological Sciences ©2003 The Japanese Pharmacological Society Full Paper Effect of TTC-909 on Cerebral Infarction Following Permanent Occlusion of the Middle Cerebral Artery in Stroke Prone Spontaneously Hypertensive Rats Yasuko Karasawa1, Hiroko Komiyama1, Shigeru Yoshida1, Noriko Hino1, Yasuhiro Katsuura2, Shiro Nakaike1 and Hiroaki Araki3,* 1Pharmacology Laboratory, Research Center, Taisho Pharmaceutical Co., Ltd., Yoshino-cho 1-403, Saitama 330-8530, Japan 2Pharmacological Research Department, Teijin Ltd., 4-3-2 Asahigaoka, Hino, Tokyo 191-8512, Japan 3Department of Hospital Pharmacy, Ehime University School of Medicine, Shiktsukawa, Shigenobu-cho, Onsen-gun, Ehime 791-0295, Japan Received November 14, 2002; Accepted January 30, 2003 Abstract. We investigated the effect of TTC-909, a drug preparation of the stable prostaglandin I2 analogue clinprost (isocarbacyclin methylester; methyl 5-{(1S,5S,6R,7R)-7- hydroxy-6-[(E)-(S)-3-hydroxy-1-octenyl] bicyclo[3.3.0]oct-2-en-3-yl} pentanoate) incorporated into lipid microspheres, on cerebral infarction 7 days after permanent occlusion of the middle cerebral artery (MCA) in stroke prone spontaneously hypertensive rats (SHRSP). Under the anesthesia, the MCA was permanently occluded above the rhinal fissure. In schedule 1, vehicle or TTC-909 was injected i.v. once daily over 7 days starting immediately after MCA occlusion. In schedule 2, vehicle or TTC-909 was infused for 3 h starting immediately after MCA occlusion. In schedule 3, vehicle or TTC-909 was infused for 3 h starting immediately after MCA occlusion followed by bolus injection once daily over 6 days. Seven days later, the infarct volume was estimated following hematoxylin and eosin staining. -

Nicotine in High Concentration Causes Contraction of Isolated Strips of Rabbit Corpus Cavernosum
Korean J Physiol Pharmacol Vol 19: 257-262, May, 2015 pISSN 1226-4512 http://dx.doi.org/10.4196/kjpp.2015.19.3.257 eISSN 2093-3827 Nicotine in High Concentration Causes Contraction of Isolated Strips of Rabbit Corpus Cavernosum Hoai Bac Nguyen1, Shin Young Lee2, Soo Hyun Park3, Jun Hyun Han4, Moo Yeol Lee3,*, and Soon Chul Myung1,* 1Advanced Urogenital Disease Research Center; Research Institute for Translational System Biomics; Department of Urology, Chung-Ang University Hospital, Seoul 156-755, 2Department of Urology, Seoul Medical Center, Seoul 131-795, 3Department of Physiology, College of Medicine, Chung-Ang University, Seoul 156-756, 4Department of Urology, Hallym University Dongtan Sacred Heart Hospital, Hwaseong 445-170, Korea It is well known that cigarette smoke can cause erectile dysfunction by affecting the penile vascular system. However, the exact effects of nicotine on the corpus cavernosum remains poorly understood. Nicotine has been reported to cause relaxation of the corpus cavernosum; it has also been reported to cause both contraction and relaxation. Therefore, high concentrations of nicotine were studied in strips from the rabbit corpus cavernosum to better understand its effects. The proximal penile corpus cavernosal strips from male rabbits weighing approximately 4 kg were used in organ bath studies. Nicotine in high concentrations (10-5∼10-4 M) produced dose-dependent contractions of the corpus cavernosal strips. The incubation with 10-5 M hexamethonium (nicotinic receptor antagonist) significantly inhibited the magnitude of the nicotine associated contractions. The nicotine-induced contractions were not only significantly inhibited by pretreatment with 10-5 M indomethacin (nonspecific cyclooxygenase inhibitor) and with 10-6 M NS-398 (selective cyclooxygenase inhibitor), but also with 10-6 M Y-27632 (Rho kinase inhibitor). -

Jp Xvii the Japanese Pharmacopoeia
JP XVII THE JAPANESE PHARMACOPOEIA SEVENTEENTH EDITION Official from April 1, 2016 English Version THE MINISTRY OF HEALTH, LABOUR AND WELFARE Notice: This English Version of the Japanese Pharmacopoeia is published for the convenience of users unfamiliar with the Japanese language. When and if any discrepancy arises between the Japanese original and its English translation, the former is authentic. The Ministry of Health, Labour and Welfare Ministerial Notification No. 64 Pursuant to Paragraph 1, Article 41 of the Law on Securing Quality, Efficacy and Safety of Products including Pharmaceuticals and Medical Devices (Law No. 145, 1960), the Japanese Pharmacopoeia (Ministerial Notification No. 65, 2011), which has been established as follows*, shall be applied on April 1, 2016. However, in the case of drugs which are listed in the Pharmacopoeia (hereinafter referred to as ``previ- ous Pharmacopoeia'') [limited to those listed in the Japanese Pharmacopoeia whose standards are changed in accordance with this notification (hereinafter referred to as ``new Pharmacopoeia'')] and have been approved as of April 1, 2016 as prescribed under Paragraph 1, Article 14 of the same law [including drugs the Minister of Health, Labour and Welfare specifies (the Ministry of Health and Welfare Ministerial Notification No. 104, 1994) as of March 31, 2016 as those exempted from marketing approval pursuant to Paragraph 1, Article 14 of the Same Law (hereinafter referred to as ``drugs exempted from approval'')], the Name and Standards established in the previous Pharmacopoeia (limited to part of the Name and Standards for the drugs concerned) may be accepted to conform to the Name and Standards established in the new Pharmacopoeia before and on September 30, 2017. -

Multidrug Treatment with Nelfinavir and Cepharanthine Against COVID-19
Supplemental Information Multidrug treatment with nelfinavir and cepharanthine against COVID-19 Hirofumi Ohashi1,2,¶, Koichi Watashi1,2,3,4*, Wakana Saso1,5.6,¶, Kaho Shionoya1,2, Shoya Iwanami7, Takatsugu Hirokawa8,9,10, Tsuyoshi Shirai11, Shigehiko Kanaya12, Yusuke Ito7, Kwang Su Kim7, Kazane Nishioka1,2, Shuji Ando13, Keisuke Ejima14, Yoshiki Koizumi15, Tomohiro Tanaka16, Shin Aoki16,17, Kouji Kuramochi2, Tadaki Suzuki18, Katsumi Maenaka19, Tetsuro Matano5,6, Masamichi Muramatsu1, Masayuki Saijo13, Kazuyuki Aihara20, Shingo Iwami4,7,21,22,23, Makoto Takeda24, Jane A. McKeating25, Takaji Wakita1 1Department of Virology II, National Institute of Infectious Diseases, Tokyo 162-8640, Japan, 2Department of Applied Biological Science, Tokyo University of Science, Noda 278-8510, Japan, 3Institute for Frontier Life and Medical Sciences, Kyoto University, Kyoto 606-8507, Japan, 4MIRAI, JST, Saitama 332-0012, Japan, 5The Institute of Medical Science, The University of Tokyo, Tokyo 108-8639, Japan, 6AIDS Research Center, National Institute of Infectious Diseases, Tokyo 162-8640, Japan, 7Department of Biology, Faculty of Sciences, Kyushu University, Fukuoka 812-8581, Japan, 8Cellular and Molecular Biotechnology Research Institute, National Institute of Advanced Industrial Science and Technology, Tokyo 135-0064, Japan, 9Division of Biomedical Science, Faculty of Medicine, University of Tsukuba, Tsukuba 305-8575, Japan, 10Transborder Medical Research Center, University of Tsukuba, Tsukuba 305-8575, Japan, 11Faculty of Bioscience, Nagahama Institute -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

The Efficacy of Ozagrel Sodium As a Rescue Or Prophylaxis in The
Research Article World Journal of Vascular Surgery Published: 16 Mar, 2018 The Efficacy of Ozagrel Sodium as a Rescue or Prophylaxis in the Perioperative Management of Thromboembolism during Coil Embolization of Ruptured and Unruptured Cerebral Aneurysms Yasuhiro Kawabata*, Norio Nakajima and Hidenori Miyake Department of Neurosurgery, Kyoto Katsura Hospital, Japan Abstract Objectives: Thromboembolism is the leading cause of neurological complication in neurointervention. We retrospectively analyzed the incidence and the outcome of thromboembolism and the efficacy of ozagrel sodium during coiling embolization of saccular cerebral aneurysm. Materials and Methods: Since 2012, we treated 103 patients with 106 aneurysms, in whom 116 procedures were performed. 35 patients were male, and 68 patients were female. The median age was 66.5 years (range 21 to 88). Sixty-two aneurysms were ruptured, and 44 aneurysms were unruptured. In a patient, coil embolization of a ruptured aneurysm and an unruptured aneurysm was performed simultaneously. Retreatment was performed in 12 patients. Ozagrel sodium, a thromboxane A2 synthase inhibitor, was administered as a rescue in 8 patients and as a prophylaxis in 17 patients prior to or during the procedure, and thrombectomy using Penumbra system® was performed in a patient. Functional outcome was assessed using Modified Rankin Scale (MRS). OPEN ACCESS Results: Thromboembolic events occurred in 27 procedures (23.3%), in 13 of which *Correspondence: thromboembolism were symptomatic. Cerebral artery occlusion was observed in 9 patients during Yasuhiro Kawabata, Department of and after the procedure, in 6 of whom early recanalization could be achieved. Procedure-related Neurosurgery, Kyoto Social Welfare permanent neurological deficit due to thromboembolism was observed in 6 patients (5.2%) Foundation, Kyoto Katsura Hospital, 17 exclusively in ruptured group; three of them had fair outcome of MRS 2-3 and other three patients Yamada Hirao-cho, Nishikyo-ku, Kyoto, had poor outcome unrelated to thromboembolism. -

A Novel JAK1 Mutant Breast Implant-Associated Anaplastic Large Cell Lymphoma Patient-Derived Xenograft Fostering Pre- Clinical Discoveries
Cancers 2019 S1 of S18 Supplementary Materials: A Novel JAK1 Mutant Breast Implant-Associated Anaplastic Large Cell Lymphoma Patient-Derived Xenograft Fostering Pre- Clinical Discoveries Danilo Fiore, Luca Vincenzo Cappelli, Paul Zumbo, Jude M. Phillip, Zhaoqi Liu, Shuhua Cheng, Liron Yoffe, Paola Ghione, Federica Di Maggio, Ahmet Dogan, Inna Khodos, Elisa de Stanchina, Joseph Casano, Clarisse Kayembe, Wayne Tam, Doron Betel, Robin Foa’, Leandro Cerchietti, Raul Rabadan, Steven Horwitz, David M. Weinstock and Giorgio Inghirami A B C Figure S1. (A) Histology micrografts on IL89 PDTX show overall similarity between T1 T3 and T7 passages (upper panels). Immunohistochemical stains with the indicated antibodies (anti-CD3, anti- CD25 and anti-CD8 [x20]) (lower panels). (B) Flow cytometry panel comprehensive of the most represented surface T-cell lymphoma markers, including: CD2, CD3, CD4, CD5, CD8, CD16, CD25, CD30, CD56, TCRab, TCRgd. IL89 PDTX passage T3 is here depicted for illustration purposes. (C) Analysis of the TCR gamma specific rearrangement clonality in IL89 diagnostic sample and correspondent PDTX after 1 and 5 passages (T1 and T5). A WT Primary p.G1097D IL89 T1 p.G1097D IL89 T5 p.G1097D IL89 cell line B Figure S2. (A) Sanger sequencing confirms the presence of the JAK1 p.G1097D mutation in IL89 PDTX samples and in the cell line, but the mutation is undetectable in the primary due to the low sensitivity of the technique. (B) Manual backtracking of mutations in the primary tumor using deep sequencing data allowed for the identification of several hits at a very low VAF compared to the PDTX-T5. A B IL89 CTRL 30 CTRL Ruxoli?nib S 20 M Ruxoli?nib A R G 10 0 1 2 3 4 5 6 7 8 9 0 1 2 3 4 1 1 1 1 1 WEEKS AFTER ENGRAFTMENT Figure S3. -

Ozagrel Hydrochloride, a Selective Thromboxane A2 Synthase Inhibitor
Tomishima et al. BMC Gastroenterology 2013, 13:21 http://www.biomedcentral.com/1471-230X/13/21 RESEARCH ARTICLE Open Access Ozagrel hydrochloride, a selective thromboxane A2 synthase inhibitor, alleviates liver injury induced by acetaminophen overdose in mice Yoshiro Tomishima1†, Yoichi Ishitsuka1*†, Naoya Matsunaga2, Minako Nagatome1, Hirokazu Furusho1, Mitsuru Irikura1, Shigehiro Ohdo2 and Tetsumi Irie1,3 Abstract Background: Overdosed acetaminophen (paracetamol, N-acetyl-p-aminophenol; APAP) causes severe liver injury. We examined the effects of ozagrel, a selective thromboxane A2 (TXA2) synthase inhibitor, on liver injury induced by APAP overdose in mice. Methods: Hepatotoxicity was induced to ICR male mice by an intraperitoneal injection with APAP (330 mg/kg). The effects of ozagrel (200 mg/kg) treatment 30 min after the APAP injection were evaluated with mortality, serum alanine aminotransferase (ALT) levels and hepatic changes, including histopathology, DNA fragmentation, mRNA expression and total glutathione contents. The impact of ozagrel (0.001-1 mg/mL) on cytochrome P450 2E1 (CYP2E1) activity in mouse hepatic microsome was examined. RLC-16 cells, a rat hepatocytes cell line, were exposed to 0.25 mM N-acetyl-p-benzoquinone imine (NAPQI), a hepatotoxic metabolite of APAP. In this model, the cytoprotective effects of ozagrel (1–100 muM) were evaluated by the WST-1 cell viability assay. Results: Ozagel treatment significantly attenuated higher mortality, elevated serum alanine aminotransferase levels, excessive hepatic centrilobular necrosis, hemorrhaging and DNA fragmentation, as well as increase in plasma 2,3-dinor thromboxane B2 levels induced by APAP injection. Ozagrel also inhibited the hepatic expression of cell death-related mRNAs induced by APAP, such as jun oncogene, FBJ osteosarcoma oncogene (fos) and C/EBP homologous protein (chop), but did not suppress B-cell lymphoma 2-like protein11 (bim) expression and hepatic total glutathione depletion. -

I in Un Torn Untuk Ta in an Atau Mana Na Anth
IIN UN TORN UNTUKTAUS IN20170304388A1 AN ATAU MANA NA ANTH ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2017/ 0304388 A1 Chen et al. ( 43 ) Pub . Date: Oct. 26 , 2017 (54 ) NEW INDICATION OF CARDIOVASCULAR A61K 31/ 549 ( 2006 . 01 ) DRUGS FOR PREPARATION OF CANCER A61K 31 /505 (2006 .01 ) INHIBITION PHARMACEUTICAL A61K 31 / 404 ( 2006 .01 ) COMPOSITION A61K 31 / 366 ( 2006 .01 ) A61K 31 /341 ( 2006 . 01 ) (71 ) Applicant : LAUNX BIOMEDICAL CO ., LTD . , A61K 31 /216 ( 2006 .01 ) Kaohsiung ( TW ) A61K 31 / 197 (2006 .01 ) A61K 31 / 196 ( 2006 .01 ) ( 72 ) Inventors : Chiu -Hung Chen , Kaohsiung ( TW ) ; A61K 45 / 06 ( 2006 . 01 ) Show -Mei Chuang, Taichung ( TW ) ; A61K 31 / 138 ( 2006 . 01) Nai- Wan Hsiao , Taichung ( TW ) ; (52 ) U . S . CI. Ruei - Yue Liang , Taichung ( TW ) ; CPC .. .. .. .. A61K 38 /05 (2013 . 01 ) ; A61K 45 / 06 Xiao - Tong Tan , Taichung ( TW ) ( 2013 .01 ) ; A61K 31 / 7056 ( 2013 .01 ) ; A61K 31/ 55 (2013 .01 ) ; A61K 31 /549 ( 2013 .01 ) ; ( 73 ) Assignee : LAUNX BIOMEDICAL CO ., LTD ., A61K 31 /505 ( 2013 . 01 ) ; A61K 31/ 138 Kaohsiung ( TW ) (2013 . 01 ) ; A61K 31/ 366 ( 2013 . 01 ) ; A61K 31/ 341 ( 2013 .01 ) ; A61K 31/ 216 ( 2013 .01 ) ; (21 ) Appl. No. : 15 /521 , 536 A61K 31 / 197 (2013 . 01 ) ; A61K 31/ 196 ( 22 ) PCT Filed : Oct. 23 , 2015 ( 2013. 01 ) ; A61K 31/ 404 ( 2013. 01 ) ( 57 ) ABSTRACT ( 86 ) PCT No .: PCT/ CN2015 /092768 A method for treating a cancer includes administering to a $ 371 ( c ) ( 1 ) , subject in need thereof a pharmaceutical composition con ( 2 ) Date : Apr. 24 , 2017 taining a therapeutically effective amount of a cardiovascu lar drug or a pharmaceutical acceptable salt thereof.