Harnessing DNA-Encoded Libraries to Discover Bioactive Small Molecules
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Prostaglandin D2 and the Role of the DP1, DP2 and TP Receptors in the Control of Airway Reflex Events
ERJ Express. Published on October 16, 2014 as doi: 10.1183/09031936.00061614 ORIGINAL ARTICLE IN PRESS | CORRECTED PROOF Prostaglandin D2 and the role of the DP1, DP2 and TP receptors in the control of airway reflex events Sarah A. Maher1, Mark A. Birrell1,2, John J. Adcock1, Michael A. Wortley1, Eric D. Dubuis1, Sara J. Bonvini1, Megan S. Grace1 and Maria G. Belvisi1,2 Affiliations: 1Respiratory Pharmacology, National Heart and Lung Institute, Faculty of Medicine, Imperial College London, London, UK. 2MRC and Asthma UK Centre in Allergic Mechanisms of Asthma, Imperial College London, London, UK. Correspondence: Maria G. Belvisi, Imperial College London, Respiratory Pharmacology, NHLI, Sir Alexander Fleming Building, Exhibition Road, London, SW7 2AZ, UK. E-mail: [email protected] ABSTRACT Prostaglandin D2 (PGD2) causes cough and levels are increased in asthma suggesting that it may contribute to symptoms. Although the prostaglandin D2 receptor 2 (DP2) is a target for numerous drug discovery programmes little is known about the actions of PGD2 on sensory nerves and cough. We used human and guinea pig bioassays, in vivo electrophysiology and a guinea pig conscious cough model to assess the effect of prostaglandin D2 receptor (DP1), DP2 and thromboxane receptor antagonism on PGD2 responses. PGD2 caused cough in a conscious guinea pig model and an increase in calcium in airway jugular ganglia. Using pharmacology and receptor-deficient mice we showed that the DP1 receptor mediates sensory nerve activation in mouse, guinea pig and human vagal afferents. In vivo, PGD2 and a DP1 receptor agonist, but not a DP2 receptor agonist, activated single airway C-fibres. -

Inverse Agonism of SQ 29,548 and Ramatroban on Thromboxane A2 Receptor
Inverse Agonism of SQ 29,548 and Ramatroban on Thromboxane A2 Receptor Raja Chakraborty1,3, Rajinder P. Bhullar1, Shyamala Dakshinamurti2,3, John Hwa4, Prashen Chelikani1,2,3* 1 Department of Oral Biology, University of Manitoba, Winnipeg, Manitoba, Canada, 2 Departments of Pediatrics, Physiology, University of Manitoba, Winnipeg, Manitoba, Canada, 3 Biology of Breathing Group- Manitoba Institute of Child Health, Winnipeg, Manitoba, Canada, 4 Department of Internal Medicine (Cardiology), Cardiovascular Research Center, Yale University School of Medicine, New Haven, Connecticut, United States of America Abstract G protein-coupled receptors (GPCRs) show some level of basal activity even in the absence of an agonist, a phenomenon referred to as constitutive activity. Such constitutive activity in GPCRs is known to have important pathophysiological roles in human disease. The thromboxane A2 receptor (TP) is a GPCR that promotes thrombosis in response to binding of the prostanoid, thromboxane A2. TP dysfunction is widely implicated in pathophysiological conditions such as bleeding disorders, hypertension and cardiovascular disease. Recently, we reported the characterization of a few constitutively active mutants (CAMs) in TP, including a genetic variant A160T. Using these CAMs as reporters, we now test the inverse agonist properties of known antagonists of TP, SQ 29,548, Ramatroban, L-670596 and Diclofenac, in HEK293T cells. Interestingly, SQ 29,548 reduced the basal activity of both, WT-TP and the CAMs while Ramatroban was able to reduce the basal activity of only the CAMs. Diclofenac and L-670596 showed no statistically significant reduction in basal activity of WT-TP or CAMs. To investigate the role of these compounds on human platelet function, we tested their effects on human megakaryocyte based system for platelet activation. -

4 Supplementary File
Supplemental Material for High-throughput screening discovers anti-fibrotic properties of Haloperidol by hindering myofibroblast activation Michael Rehman1, Simone Vodret1, Luca Braga2, Corrado Guarnaccia3, Fulvio Celsi4, Giulia Rossetti5, Valentina Martinelli2, Tiziana Battini1, Carlin Long2, Kristina Vukusic1, Tea Kocijan1, Chiara Collesi2,6, Nadja Ring1, Natasa Skoko3, Mauro Giacca2,6, Giannino Del Sal7,8, Marco Confalonieri6, Marcello Raspa9, Alessandro Marcello10, Michael P. Myers11, Sergio Crovella3, Paolo Carloni5, Serena Zacchigna1,6 1Cardiovascular Biology, 2Molecular Medicine, 3Biotechnology Development, 10Molecular Virology, and 11Protein Networks Laboratories, International Centre for Genetic Engineering and Biotechnology (ICGEB), Padriciano, 34149, Trieste, Italy 4Institute for Maternal and Child Health, IRCCS "Burlo Garofolo", Trieste, Italy 5Computational Biomedicine Section, Institute of Advanced Simulation IAS-5 and Institute of Neuroscience and Medicine INM-9, Forschungszentrum Jülich GmbH, 52425, Jülich, Germany 6Department of Medical, Surgical and Health Sciences, University of Trieste, 34149 Trieste, Italy 7National Laboratory CIB, Area Science Park Padriciano, Trieste, 34149, Italy 8Department of Life Sciences, University of Trieste, Trieste, 34127, Italy 9Consiglio Nazionale delle Ricerche (IBCN), CNR-Campus International Development (EMMA- INFRAFRONTIER-IMPC), Rome, Italy This PDF file includes: Supplementary Methods Supplementary References Supplementary Figures with legends 1 – 18 Supplementary Tables with legends 1 – 5 Supplementary Movie legends 1, 2 Supplementary Methods Cell culture Primary murine fibroblasts were isolated from skin, lung, kidney and hearts of adult CD1, C57BL/6 or aSMA-RFP/COLL-EGFP mice (1) by mechanical and enzymatic tissue digestion. Briefly, tissue was chopped in small chunks that were digested using a mixture of enzymes (Miltenyi Biotec, 130- 098-305) for 1 hour at 37°C with mechanical dissociation followed by filtration through a 70 µm cell strainer and centrifugation. -
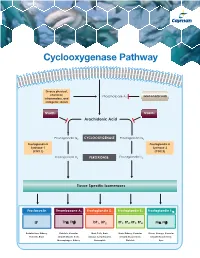
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Cr
Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Crizotinib (PF-02341066) 1 4 55 Docetaxel 1 5 98 Anastrozole 1 6 25 Cladribine 1 7 23 Methotrexate 1 8 -187 Letrozole 1 9 65 Entecavir Hydrate 1 10 48 Roxadustat (FG-4592) 1 11 19 Imatinib Mesylate (STI571) 1 12 0 Sunitinib Malate 1 13 34 Vismodegib (GDC-0449) 1 14 64 Paclitaxel 1 15 89 Aprepitant 1 16 94 Decitabine 1 17 -79 Bendamustine HCl 1 18 19 Temozolomide 1 19 -111 Nepafenac 1 20 24 Nintedanib (BIBF 1120) 1 21 -43 Lapatinib (GW-572016) Ditosylate 1 22 88 Temsirolimus (CCI-779, NSC 683864) 1 23 96 Belinostat (PXD101) 1 24 46 Capecitabine 1 25 19 Bicalutamide 1 26 83 Dutasteride 1 27 68 Epirubicin HCl 1 28 -59 Tamoxifen 1 29 30 Rufinamide 1 30 96 Afatinib (BIBW2992) 1 31 -54 Lenalidomide (CC-5013) 1 32 19 Vorinostat (SAHA, MK0683) 1 33 38 Rucaparib (AG-014699,PF-01367338) phosphate1 34 14 Lenvatinib (E7080) 1 35 80 Fulvestrant 1 36 76 Melatonin 1 37 15 Etoposide 1 38 -69 Vincristine sulfate 1 39 61 Posaconazole 1 40 97 Bortezomib (PS-341) 1 41 71 Panobinostat (LBH589) 1 42 41 Entinostat (MS-275) 1 43 26 Cabozantinib (XL184, BMS-907351) 1 44 79 Valproic acid sodium salt (Sodium valproate) 1 45 7 Raltitrexed 1 46 39 Bisoprolol fumarate 1 47 -23 Raloxifene HCl 1 48 97 Agomelatine 1 49 35 Prasugrel 1 50 -24 Bosutinib (SKI-606) 1 51 85 Nilotinib (AMN-107) 1 52 99 Enzastaurin (LY317615) 1 53 -12 Everolimus (RAD001) 1 54 94 Regorafenib (BAY 73-4506) 1 55 24 Thalidomide 1 56 40 Tivozanib (AV-951) 1 57 86 Fludarabine -

Clinical Trial Protocol: BPS-314D-MR-PAH-302
Clinical Trial Protocol: BPS-314d-MR-PAH-302 Study Title: A multicenter, double-blind, randomized, placebo-controlled, Phase 3 study to assess the efficacy and safety of oral BPS-314d-MR added-on to treprostinil, inhaled (Tyvaso®) in subjects with pulmonary arterial hypertension Study Number: BPS-314d- MR-PAH-302 Study Phase: 3 Product Name: Beraprost Sodium 314d Modified Release IND Number: 111,729 Indication: Treatment of Pulmonary Arterial Hypertension Investigators: Multicenter Sponsor: Lung Biotechnology Inc. Sponsor Contact: Medical Monitor: Date Original Protocol: 16 April 2013 Amendment 1: 17 December 2013 Amendment 2 15 October 2014 GCP Statement: This study will be conducted in compliance with the protocol, Good Clinical Practice (GCP) and applicable regulatory requirements. Confidentiality Statement The concepts and information contained herein are confidential and proprietary and shall not be disclosed in whole or part without the express written consent of Lung Biotechnology Inc. © 2014 Lung Biotechnology Inc. Beraprost Sodium 314d Modified Release Lung Biotechnology Inc. Clinical Trial Protocol: BPS-314-d-MR-PAH 302 15 October 2014 LIST OF CONTACTS Study Sponsor: Lung Biotechnology Inc. 1040 Spring Street Silver Spring, Maryland 20910 United States Phone: 301-608-9292 Fax: 301-589-0855 Sponsor Contact: Medical Monitor: SAE Reporting: CONFIDENTIAL Page 2 of 75 Beraprost Sodium 314d Modified Release Lung Biotechnology Inc. Clinical Trial Protocol: BPS-314-d-MR-PAH 302 15 October 2014 SYNOPSIS Sponsor: Lung Biotechnology Inc. -

Effect of Prostanoids on Human Platelet Function: an Overview
International Journal of Molecular Sciences Review Effect of Prostanoids on Human Platelet Function: An Overview Steffen Braune, Jan-Heiner Küpper and Friedrich Jung * Institute of Biotechnology, Molecular Cell Biology, Brandenburg University of Technology, 01968 Senftenberg, Germany; steff[email protected] (S.B.); [email protected] (J.-H.K.) * Correspondence: [email protected] Received: 23 October 2020; Accepted: 23 November 2020; Published: 27 November 2020 Abstract: Prostanoids are bioactive lipid mediators and take part in many physiological and pathophysiological processes in practically every organ, tissue and cell, including the vascular, renal, gastrointestinal and reproductive systems. In this review, we focus on their influence on platelets, which are key elements in thrombosis and hemostasis. The function of platelets is influenced by mediators in the blood and the vascular wall. Activated platelets aggregate and release bioactive substances, thereby activating further neighbored platelets, which finally can lead to the formation of thrombi. Prostanoids regulate the function of blood platelets by both activating or inhibiting and so are involved in hemostasis. Each prostanoid has a unique activity profile and, thus, a specific profile of action. This article reviews the effects of the following prostanoids: prostaglandin-D2 (PGD2), prostaglandin-E1, -E2 and E3 (PGE1, PGE2, PGE3), prostaglandin F2α (PGF2α), prostacyclin (PGI2) and thromboxane-A2 (TXA2) on platelet activation and aggregation via their respective receptors. Keywords: prostacyclin; thromboxane; prostaglandin; platelets 1. Introduction Hemostasis is a complex process that requires the interplay of multiple physiological pathways. Cellular and molecular mechanisms interact to stop bleedings of injured blood vessels or to seal denuded sub-endothelium with localized clot formation (Figure1). -

Effect of TTC-909 on Cerebral Infarction Following Permanent Occlusion of the Middle Cerebral Artery in Stroke Prone Spontaneously Hypertensive Rats
J Pharmacol Sci 91, 305 – 312 (2003) Journal of Pharmacological Sciences ©2003 The Japanese Pharmacological Society Full Paper Effect of TTC-909 on Cerebral Infarction Following Permanent Occlusion of the Middle Cerebral Artery in Stroke Prone Spontaneously Hypertensive Rats Yasuko Karasawa1, Hiroko Komiyama1, Shigeru Yoshida1, Noriko Hino1, Yasuhiro Katsuura2, Shiro Nakaike1 and Hiroaki Araki3,* 1Pharmacology Laboratory, Research Center, Taisho Pharmaceutical Co., Ltd., Yoshino-cho 1-403, Saitama 330-8530, Japan 2Pharmacological Research Department, Teijin Ltd., 4-3-2 Asahigaoka, Hino, Tokyo 191-8512, Japan 3Department of Hospital Pharmacy, Ehime University School of Medicine, Shiktsukawa, Shigenobu-cho, Onsen-gun, Ehime 791-0295, Japan Received November 14, 2002; Accepted January 30, 2003 Abstract. We investigated the effect of TTC-909, a drug preparation of the stable prostaglandin I2 analogue clinprost (isocarbacyclin methylester; methyl 5-{(1S,5S,6R,7R)-7- hydroxy-6-[(E)-(S)-3-hydroxy-1-octenyl] bicyclo[3.3.0]oct-2-en-3-yl} pentanoate) incorporated into lipid microspheres, on cerebral infarction 7 days after permanent occlusion of the middle cerebral artery (MCA) in stroke prone spontaneously hypertensive rats (SHRSP). Under the anesthesia, the MCA was permanently occluded above the rhinal fissure. In schedule 1, vehicle or TTC-909 was injected i.v. once daily over 7 days starting immediately after MCA occlusion. In schedule 2, vehicle or TTC-909 was infused for 3 h starting immediately after MCA occlusion. In schedule 3, vehicle or TTC-909 was infused for 3 h starting immediately after MCA occlusion followed by bolus injection once daily over 6 days. Seven days later, the infarct volume was estimated following hematoxylin and eosin staining. -

Antiplatelet Effects of Prostacyclin Analogues: Which One to Choose in Case of Thrombosis Or Bleeding? Sylwester P
INTERVENTIONAL CARDIOLOGY Cardiology Journal 20XX, Vol. XX, No. X, XXX–XXX DOI: 10.5603/CJ.a2020.0164 Copyright © 20XX Via Medica REVIEW ARTICLE ISSN 1897–5593 eISSN 1898–018X Antiplatelet effects of prostacyclin analogues: Which one to choose in case of thrombosis or bleeding? Sylwester P. Rogula1*, Hubert M. Mutwil1*, Aleksandra Gąsecka1, Marcin Kurzyna2, Krzysztof J. Filipiak1 11st Chair and Department of Cardiology, Medical University of Warsaw, Poland 2Department of Pulmonary Circulation, Thromboembolic Diseases and Cardiology, Center of Postgraduate Education Medical, European Health Center Otwock, Poland Abstract Prostacyclin and analogues are successfully used in the treatment of pulmonary arterial hypertension (PAH) due to their vasodilatory effect on pulmonary arteries. Besides vasodilatory effect, prostacyclin analogues inhibit platelets, but their antiplatelet effect is not thoroughly established. The antiplatelet effect of prostacyclin analogues may be beneficial in case of increased risk of thromboembolic events, or undesirable in case of increased risk of bleeding. Since prostacyclin and analogues differ regarding their potency and form of administration, they might also inhibit platelets to a different extent. This review summarizes the recent evidence on the antiplatelet effects of prostacyclin and analogue in the treatment of PAH, this is important to consider when choosing the optimal treatment regimen in tailoring to an individual patients’ needs. (Cardiol J 20XX; XX, X: xx–xx) Key words: prostacyclin analogues, pulmonary arterial hypertension, platelets, antiplatelet effect, thrombosis, bleeding Introduction tiproliferative effects [4]. The main indication for PGI2 and analogues is advanced pulmonary arterial Since 1935 when prostaglandin was isolated hypertension (PAH) and peripheral vascular disor- for the first time [1], many scientists have focused ders [5]. -

Prostacyclin Therapies for the Treatment of Pulmonary Arterial Hypertension
Eur Respir J 2008; 31: 891–901 DOI: 10.1183/09031936.00097107 CopyrightßERS Journals Ltd 2008 SERIES ‘‘PULMONARY HYPERTENSION: BASIC CONCEPTS FOR PRACTICAL MANAGEMENT’’ Edited by M.M. Hoeper and A.T. Dinh-Xuan Number 2 in this Series Prostacyclin therapies for the treatment of pulmonary arterial hypertension M. Gomberg-Maitland* and H. Olschewski# ABSTRACT: Prostacyclin and its analogues (prostanoids) are potent vasodilators and possess AFFILIATIONS antithrombotic, antiproliferative and anti-inflammatory properties. Pulmonary hypertension (PH) *Dept of Cardiology, University of Chicago Hospitals, Chicago, IL, USA. is associated with vasoconstriction, thrombosis and proliferation, and the lack of endogenous #Dept of Pulmonology, Medical prostacyclin may considerably contribute to this condition. This supports a strong rationale for University Graz, Graz, Austria. prostanoid use as therapy for this disease. The first experiences of prostanoid therapy in PH patients were published in 1980. CORRESPONDENCE H. Olschewski Epoprostenol, a synthetic analogue of prostacyclin, and the chemically stable analogues Dept of Pulmonology iloprost, beraprost and treprostinil were tested in randomised controlled trials. The biological Medical University Graz actions are mainly mediated by activation of specific receptors of the target cells; however, new Auenbruggerplatz 20 data suggest effects on additional intracellular pathways. In the USA and some European Graz 8010 Austria countries, intravenous infusion of epoprostenol and treprostinil, as well as subcutaneous infusion Fax: 43 3163853578 of treprostinil and inhalation of iloprost, have been approved for therapy of pulmonary arterial E-mail: horst.olschewski@ hypertension. Iloprost infusion and beraprost tablets have been approved in few other countries. meduni-graz.at Ongoing clinical studies investigate oral treprostinil, inhaled treprostinil and the combination of Received: inhaled iloprost and sildenafil in pulmonary arterial hypertension. -

Nicotine in High Concentration Causes Contraction of Isolated Strips of Rabbit Corpus Cavernosum
Korean J Physiol Pharmacol Vol 19: 257-262, May, 2015 pISSN 1226-4512 http://dx.doi.org/10.4196/kjpp.2015.19.3.257 eISSN 2093-3827 Nicotine in High Concentration Causes Contraction of Isolated Strips of Rabbit Corpus Cavernosum Hoai Bac Nguyen1, Shin Young Lee2, Soo Hyun Park3, Jun Hyun Han4, Moo Yeol Lee3,*, and Soon Chul Myung1,* 1Advanced Urogenital Disease Research Center; Research Institute for Translational System Biomics; Department of Urology, Chung-Ang University Hospital, Seoul 156-755, 2Department of Urology, Seoul Medical Center, Seoul 131-795, 3Department of Physiology, College of Medicine, Chung-Ang University, Seoul 156-756, 4Department of Urology, Hallym University Dongtan Sacred Heart Hospital, Hwaseong 445-170, Korea It is well known that cigarette smoke can cause erectile dysfunction by affecting the penile vascular system. However, the exact effects of nicotine on the corpus cavernosum remains poorly understood. Nicotine has been reported to cause relaxation of the corpus cavernosum; it has also been reported to cause both contraction and relaxation. Therefore, high concentrations of nicotine were studied in strips from the rabbit corpus cavernosum to better understand its effects. The proximal penile corpus cavernosal strips from male rabbits weighing approximately 4 kg were used in organ bath studies. Nicotine in high concentrations (10-5∼10-4 M) produced dose-dependent contractions of the corpus cavernosal strips. The incubation with 10-5 M hexamethonium (nicotinic receptor antagonist) significantly inhibited the magnitude of the nicotine associated contractions. The nicotine-induced contractions were not only significantly inhibited by pretreatment with 10-5 M indomethacin (nonspecific cyclooxygenase inhibitor) and with 10-6 M NS-398 (selective cyclooxygenase inhibitor), but also with 10-6 M Y-27632 (Rho kinase inhibitor). -

Jp Xvii the Japanese Pharmacopoeia
JP XVII THE JAPANESE PHARMACOPOEIA SEVENTEENTH EDITION Official from April 1, 2016 English Version THE MINISTRY OF HEALTH, LABOUR AND WELFARE Notice: This English Version of the Japanese Pharmacopoeia is published for the convenience of users unfamiliar with the Japanese language. When and if any discrepancy arises between the Japanese original and its English translation, the former is authentic. The Ministry of Health, Labour and Welfare Ministerial Notification No. 64 Pursuant to Paragraph 1, Article 41 of the Law on Securing Quality, Efficacy and Safety of Products including Pharmaceuticals and Medical Devices (Law No. 145, 1960), the Japanese Pharmacopoeia (Ministerial Notification No. 65, 2011), which has been established as follows*, shall be applied on April 1, 2016. However, in the case of drugs which are listed in the Pharmacopoeia (hereinafter referred to as ``previ- ous Pharmacopoeia'') [limited to those listed in the Japanese Pharmacopoeia whose standards are changed in accordance with this notification (hereinafter referred to as ``new Pharmacopoeia'')] and have been approved as of April 1, 2016 as prescribed under Paragraph 1, Article 14 of the same law [including drugs the Minister of Health, Labour and Welfare specifies (the Ministry of Health and Welfare Ministerial Notification No. 104, 1994) as of March 31, 2016 as those exempted from marketing approval pursuant to Paragraph 1, Article 14 of the Same Law (hereinafter referred to as ``drugs exempted from approval'')], the Name and Standards established in the previous Pharmacopoeia (limited to part of the Name and Standards for the drugs concerned) may be accepted to conform to the Name and Standards established in the new Pharmacopoeia before and on September 30, 2017.