Supplement Ii to the Japanese Pharmacopoeia Seventeenth Edition
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
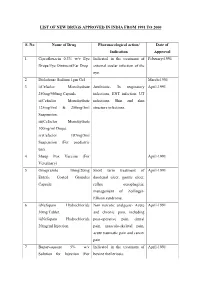
List of New Drugs Approved in India from 1991 to 2000
LIST OF NEW DRUGS APPROVED IN INDIA FROM 1991 TO 2000 S. No Name of Drug Pharmacological action/ Date of Indication Approval 1 Ciprofloxacin 0.3% w/v Eye Indicated in the treatment of February-1991 Drops/Eye Ointment/Ear Drop external ocular infection of the eye. 2 Diclofenac Sodium 1gm Gel March-1991 3 i)Cefaclor Monohydrate Antibiotic- In respiratory April-1991 250mg/500mg Capsule. infections, ENT infection, UT ii)Cefaclor Monohydrate infections, Skin and skin 125mg/5ml & 250mg/5ml structure infections. Suspension. iii)Cefaclor Monohydrate 100mg/ml Drops. iv)Cefaclor 187mg/5ml Suspension (For paediatric use). 4 Sheep Pox Vaccine (For April-1991 Veterinary) 5 Omeprazole 10mg/20mg Short term treatment of April-1991 Enteric Coated Granules duodenal ulcer, gastric ulcer, Capsule reflux oesophagitis, management of Zollinger- Ellison syndrome. 6 i)Nefopam Hydrochloride Non narcotic analgesic- Acute April-1991 30mg Tablet. and chronic pain, including ii)Nefopam Hydrochloride post-operative pain, dental 20mg/ml Injection. pain, musculo-skeletal pain, acute traumatic pain and cancer pain. 7 Buparvaquone 5% w/v Indicated in the treatment of April-1991 Solution for Injection (For bovine theileriosis. Veterinary) 8 i)Kitotifen Fumerate 1mg Anti asthmatic drug- Indicated May-1991 Tablet in prophylactic treatment of ii)Kitotifen Fumerate Syrup bronchial asthma, symptomatic iii)Ketotifen Fumerate Nasal improvement of allergic Drops conditions including rhinitis and conjunctivitis. 9 i)Pefloxacin Mesylate Antibacterial- In the treatment May-1991 Dihydrate 400mg Film Coated of severe infection in adults Tablet caused by sensitive ii)Pefloxacin Mesylate microorganism (gram -ve Dihydrate 400mg/5ml Injection pathogens and staphylococci). iii)Pefloxacin Mesylate Dihydrate 400mg I.V Bottles of 100ml/200ml 10 Ofloxacin 100mg/50ml & Indicated in RTI, UTI, May-1991 200mg/100ml vial Infusion gynaecological infection, skin/soft lesion infection. -

WHO Drug Information Vol. 12, No. 3, 1998
WHO DRUG INFORMATION VOLUME 12 NUMBER 3 • 1998 RECOMMENDED INN LIST 40 INTERNATIONAL NONPROPRIETARY NAMES FOR PHARMACEUTICAL SUBSTANCES WORLD HEALTH ORGANIZATION • GENEVA Volume 12, Number 3, 1998 World Health Organization, Geneva WHO Drug Information Contents Seratrodast and hepatic dysfunction 146 Meloxicam safety similar to other NSAIDs 147 Proxibarbal withdrawn from the market 147 General Policy Issues Cholestin an unapproved drug 147 Vigabatrin and visual defects 147 Starting materials for pharmaceutical products: safety concerns 129 Glycerol contaminated with diethylene glycol 129 ATC/DDD Classification (final) 148 Pharmaceutical excipients: certificates of analysis and vendor qualification 130 ATC/DDD Classification Quality assurance and supply of starting (temporary) 150 materials 132 Implementation of vendor certification 134 Control and safe trade in starting materials Essential Drugs for pharmaceuticals: recommendations 134 WHO Model Formulary: Immunosuppressives, antineoplastics and drugs used in palliative care Reports on Individual Drugs Immunosuppresive drugs 153 Tamoxifen in the prevention and treatment Azathioprine 153 of breast cancer 136 Ciclosporin 154 Selective serotonin re-uptake inhibitors and Cytotoxic drugs 154 withdrawal reactions 136 Asparaginase 157 Triclabendazole and fascioliasis 138 Bleomycin 157 Calcium folinate 157 Chlormethine 158 Current Topics Cisplatin 158 Reverse transcriptase activity in vaccines 140 Cyclophosphamide 158 Consumer protection and herbal remedies 141 Cytarabine 159 Indiscriminate antibiotic -

Aldrich FT-IR Collection Edition I Library
Aldrich FT-IR Collection Edition I Library Library Listing – 10,505 spectra This library is the original FT-IR spectral collection from Aldrich. It includes a wide variety of pure chemical compounds found in the Aldrich Handbook of Fine Chemicals. The Aldrich Collection of FT-IR Spectra Edition I library contains spectra of 10,505 pure compounds and is a subset of the Aldrich Collection of FT-IR Spectra Edition II library. All spectra were acquired by Sigma-Aldrich Co. and were processed by Thermo Fisher Scientific. Eight smaller Aldrich Material Specific Sub-Libraries are also available. Aldrich FT-IR Collection Edition I Index Compound Name Index Compound Name 3515 ((1R)-(ENDO,ANTI))-(+)-3- 928 (+)-LIMONENE OXIDE, 97%, BROMOCAMPHOR-8- SULFONIC MIXTURE OF CIS AND TRANS ACID, AMMONIUM SALT 209 (+)-LONGIFOLENE, 98+% 1708 ((1R)-ENDO)-(+)-3- 2283 (+)-MURAMIC ACID HYDRATE, BROMOCAMPHOR, 98% 98% 3516 ((1S)-(ENDO,ANTI))-(-)-3- 2966 (+)-N,N'- BROMOCAMPHOR-8- SULFONIC DIALLYLTARTARDIAMIDE, 99+% ACID, AMMONIUM SALT 2976 (+)-N-ACETYLMURAMIC ACID, 644 ((1S)-ENDO)-(-)-BORNEOL, 99% 97% 9587 (+)-11ALPHA-HYDROXY-17ALPHA- 965 (+)-NOE-LACTOL DIMER, 99+% METHYLTESTOSTERONE 5127 (+)-P-BROMOTETRAMISOLE 9590 (+)-11ALPHA- OXALATE, 99% HYDROXYPROGESTERONE, 95% 661 (+)-P-MENTH-1-EN-9-OL, 97%, 9588 (+)-17-METHYLTESTOSTERONE, MIXTURE OF ISOMERS 99% 730 (+)-PERSEITOL 8681 (+)-2'-DEOXYURIDINE, 99+% 7913 (+)-PILOCARPINE 7591 (+)-2,3-O-ISOPROPYLIDENE-2,3- HYDROCHLORIDE, 99% DIHYDROXY- 1,4- 5844 (+)-RUTIN HYDRATE, 95% BIS(DIPHENYLPHOSPHINO)BUT 9571 (+)-STIGMASTANOL -

(12) Patent Application Publication (10) Pub. No.: US 2006/0110428A1 De Juan Et Al
US 200601 10428A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2006/0110428A1 de Juan et al. (43) Pub. Date: May 25, 2006 (54) METHODS AND DEVICES FOR THE Publication Classification TREATMENT OF OCULAR CONDITIONS (51) Int. Cl. (76) Inventors: Eugene de Juan, LaCanada, CA (US); A6F 2/00 (2006.01) Signe E. Varner, Los Angeles, CA (52) U.S. Cl. .............................................................. 424/427 (US); Laurie R. Lawin, New Brighton, MN (US) (57) ABSTRACT Correspondence Address: Featured is a method for instilling one or more bioactive SCOTT PRIBNOW agents into ocular tissue within an eye of a patient for the Kagan Binder, PLLC treatment of an ocular condition, the method comprising Suite 200 concurrently using at least two of the following bioactive 221 Main Street North agent delivery methods (A)-(C): Stillwater, MN 55082 (US) (A) implanting a Sustained release delivery device com (21) Appl. No.: 11/175,850 prising one or more bioactive agents in a posterior region of the eye so that it delivers the one or more (22) Filed: Jul. 5, 2005 bioactive agents into the vitreous humor of the eye; (B) instilling (e.g., injecting or implanting) one or more Related U.S. Application Data bioactive agents Subretinally; and (60) Provisional application No. 60/585,236, filed on Jul. (C) instilling (e.g., injecting or delivering by ocular ion 2, 2004. Provisional application No. 60/669,701, filed tophoresis) one or more bioactive agents into the Vit on Apr. 8, 2005. reous humor of the eye. Patent Application Publication May 25, 2006 Sheet 1 of 22 US 2006/0110428A1 R 2 2 C.6 Fig. -

Oregon Department of Human Services HEALTH EFFECTS INFORMATION
Oregon Department of Human Services Office of Environmental Public Health (503) 731-4030 Emergency 800 NE Oregon Street #604 (971) 673-0405 Portland, OR 97232-2162 (971) 673-0457 FAX (971) 673-0372 TTY-Nonvoice TECHNICAL BULLETIN HEALTH EFFECTS INFORMATION Prepared by: Department of Human Services ENVIRONMENTAL TOXICOLOGY SECTION Office of Environmental Public Health OCTOBER, 1998 CALCIUM CARBONATE "lime, limewater” For More Information Contact: Environmental Toxicology Section (971) 673-0440 Drinking Water Section (971) 673-0405 Technical Bulletin - Health Effects Information CALCIUM CARBONATE, "lime, limewater@ Page 2 SYNONYMS: Lime, ground limestone, dolomite, sugar lime, oyster shell, coral shell, marble dust, calcite, whiting, marl dust, putty dust CHEMICAL AND PHYSICAL PROPERTIES: - Molecular Formula: CaCO3 - White solid, crystals or powder, may draw moisture from the air and become damp on exposure - Odorless, chalky, flat, sweetish flavor (Do not confuse with "anhydrous lime" which is a special form of calcium hydroxide, an extremely caustic, dangerous product. Direct contact with it is immediately injurious to skin, eyes, intestinal tract and respiratory system.) WHERE DOES CALCIUM CARBONATE COME FROM? Calcium carbonate can be mined from the earth in solid form or it may be extracted from seawater or other brines by industrial processes. Natural shells, bones and chalk are composed predominantly of calcium carbonate. WHAT ARE THE PRINCIPLE USES OF CALCIUM CARBONATE? Calcium carbonate is an important ingredient of many household products. It is used as a whitening agent in paints, soaps, art products, paper, polishes, putty products and cement. It is used as a filler and whitener in many cosmetic products including mouth washes, creams, pastes, powders and lotions. -

Ocean Acidification and Oceanic Carbon Cycling 13
Ocean Acidification and Oceanic Carbon Cycling 13 Dieter A. Wolf-Gladrow and Bjo¨rn Rost Contents Atmospheric CO2 ............................................................................... 104 Air-Sea CO2 Exchange and Ocean Carbonate Chemistry ..................................... 104 Rate of Surface Ocean Acidification and Regional Differences .. ........................... 104 The Physical Carbon Pump and Climate Change .............................................. 105 Impact of Ocean Acidification on Marine Organisms and Ecosystems ....................... 106 The Biological Carbon Pump and Climate Change ............................................ 107 Take-Home Message ............................................................................ 108 References ....................................................................................... 109 Abstract The concentration of atmospheric CO2 is increasing due to emissions from burning of fossil fuels and changes in land use. Part of this “anthropogenic CO2” invades the oceans causing a decrease of seawater pH; this process is called “ocean acidification.” The lowered pH, but also the concomitant changes in other properties of the carbonate system, affects marine life and the cycling of carbon in the ocean. Keywords Anthropogenic CO2 • Seawater acidity • Saturation state • Climate change • Physical carbon pump • Global warming • Biological carbon pumps • Phyto- plankton • Primary production • Calcification D.A. Wolf-Gladrow (*) • B. Rost Alfred Wegener Institute, Helmholtz Centre -

Dorset Medicines Advisory Group
DORSET CARDIOLOGY WORKING GROUP GUIDELINE FOR CALCIUM CHANNEL BLOCKERS IN HYPERTENSION SUMMARY The pan-Dorset cardiology working group continues to recommend the use of amlodipine (a third generation dihydropyridine calcium-channel blocker) as first choice calcium channel blocker on the pan-Dorset formulary for hypertension. Lercanidipine is second choice, lacidipine third choice and felodipine is fourth choice. This is due to preferable side effect profiles in terms of ankle oedema and relative costs of the preparations. Note: where angina is the primary indication or is a co-morbidity prescribers must check against the specific product characteristics (SPC) for an individual drug to confirm this is a licensed indication. N.B. Lacidipine and lercandipine are only licensed for use in hypertension. Chapter 02.06.02 CCBs section of the Formulary has undergone an evidence-based review. A comprehensive literature search was carried out on NHS Evidence, Medline, EMBASE, Cochrane Database, and UK Duets. This was for recent reviews or meta-analyses on calcium channel blockers from 2009 onwards (comparative efficacy and side effects) and randomised controlled trials (RCTs). REVIEW BACKGROUND Very little good quality evidence exists. No reviews, meta-analyses or RCTs were found covering all calcium channel blockers currently on the formulary. Another limitation was difficulty obtaining full text original papers for some of the references therefore having to use those from more obscure journals instead. Some discrepancies exist between classification of generations of dihydropyridine CCBs, depending upon the year of publication of the reference/authors’ interpretation. Dihydropyridine (DHP) CCBs tend to be more potent vasodilators than non-dihydropyridine (non-DHP) CCBs (diltiazem, verapamil), but the latter have greater inotropic effects. -

Calcium Channel Blocker As a Drug Candidate for the Treatment of Generalised Epilepsies
UNIVERSITAT DE BARCELONA Faculty of Pharmacy and Food Sciences Calcium channel blocker as a drug candidate for the treatment of generalised epilepsies Final degree project Author: Janire Sanz Sevilla Bachelor's degree in Pharmacy Primary field: Organic Chemistry, Pharmacology and Therapeutics Secondary field: Physiology, Pathophysiology and Molecular Biology March 2019 This work is licensed under a Creative Commons license ABBREVIATIONS AED antiepileptic drug AMPA α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid ANNA-1 antineuronal nuclear antibody 1 BBB blood-brain barrier Bn benzyl BnBr benzyl bromide BnNCO benzyl isocyanate Boc tert-butoxycarbonyl Bu4NBr tetrabutylammonium bromide Ca+2 calcium ion CACNA1 calcium channel voltage-dependent gene cAMP cyclic adenosine monophosphate CCB calcium channel blocker cGMP cyclic guanosine monophosphate CH3CN acetonitrile Cl- chlorine ion Cmax maximum concentration CMV cytomegalovirus CTScan computed axial tomography DCM dichloromethane DIPEA N,N-diisopropylethylamine DMF dimethylformamide DMPK drug metabolism and pharmacokinetics DNET dysembryoplastic neuroepithelial tumours EEG electroencephalogram EPSP excitatory post-synaptic potential FDA food and drug administration Fe iron FLIPR fluorescence imaging plate reader fMRI functional magnetic resonance imaging GABA γ-amino-α-hydroxybutyric acid GAD65 glutamic acid decarboxylase 65 GAERS generalised absence epilepsy rat of Strasbourg GluR5 kainate receptor GTC generalised tonic-clonic H+ hydrogen ion H2 hydrogen H2O dihydrogen dioxide (water) -

Customs Tariff - Schedule
CUSTOMS TARIFF - SCHEDULE 99 - i Chapter 99 SPECIAL CLASSIFICATION PROVISIONS - COMMERCIAL Notes. 1. The provisions of this Chapter are not subject to the rule of specificity in General Interpretative Rule 3 (a). 2. Goods which may be classified under the provisions of Chapter 99, if also eligible for classification under the provisions of Chapter 98, shall be classified in Chapter 98. 3. Goods may be classified under a tariff item in this Chapter and be entitled to the Most-Favoured-Nation Tariff or a preferential tariff rate of customs duty under this Chapter that applies to those goods according to the tariff treatment applicable to their country of origin only after classification under a tariff item in Chapters 1 to 97 has been determined and the conditions of any Chapter 99 provision and any applicable regulations or orders in relation thereto have been met. 4. The words and expressions used in this Chapter have the same meaning as in Chapters 1 to 97. Issued January 1, 2020 99 - 1 CUSTOMS TARIFF - SCHEDULE Tariff Unit of MFN Applicable SS Description of Goods Item Meas. Tariff Preferential Tariffs 9901.00.00 Articles and materials for use in the manufacture or repair of the Free CCCT, LDCT, GPT, UST, following to be employed in commercial fishing or the commercial MT, MUST, CIAT, CT, harvesting of marine plants: CRT, IT, NT, SLT, PT, COLT, JT, PAT, HNT, Artificial bait; KRT, CEUT, UAT, CPTPT: Free Carapace measures; Cordage, fishing lines (including marlines), rope and twine, of a circumference not exceeding 38 mm; Devices for keeping nets open; Fish hooks; Fishing nets and netting; Jiggers; Line floats; Lobster traps; Lures; Marker buoys of any material excluding wood; Net floats; Scallop drag nets; Spat collectors and collector holders; Swivels. -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

(19) United States (12) Patent Application Publication (10) Pub
US 20050181041A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2005/0181041 A1 Goldman (43) Pub. Date: Aug. 18, 2005 (54) METHOD OF PREPARATION OF MIXED Related US. Application Data PHASE CO-CRYSTALS WITH ACTIVE AGENTS (60) Provisional application No. 60/528,232, ?led on Dec. 9, 2003. Provisional application No. 60/559,862, ?led (75) Inventor: David Goldman, Portland, CT (US) on Apr. 6, 2004. Correspondence Address: Publication Classi?cation LEYDIG VOIT & MAYER, LTD (51) Int. Cl.7 ....................... .. A61K 31/56; A61K 38/00; TWO PRUDENTIAL PLAZA, SUITE 4900 A61K 9/64 180 NORTH STETSON AVENUE (52) US. Cl. ............................ .. 424/456; 514/179; 514/2; CHICAGO, IL 60601-6780 (US) 514/221 (73) Assignee: MedCrystalForms, LLC, Hunt Valley, (57) ABSTRACT MD This invention pertains to a method of preparing mixed phase co-crystals of active agents With one or more materials (21) Appl. No.: 11/008,034 that alloWs the modi?cation of the active agent to a neW physical/crystal form With unique properties useful for the delivery of the active agent, as Well as compositions com (22) Filed: Dec. 9, 2004 prising the mixed phase co-crystals. Patent Application Publication Aug. 18, 2005 Sheet 1 0f 8 US 2005/0181041 A1 FIG. 1a 214.70°C z.m."m.n... 206.98°C n..0ao 142 OJ/g as:20m=3: -0.8 -1.0 40 90 1:10 2110 Temperture (°C) FIG. 1b 0.01 as:22“.Km: 217 095 24221.4 39Jmum/Q -0.8 35 155 255 255 Temperture (°C) Patent Application Publication Aug. -

Tall Man Lettering List REPORT DECEMBER 2013 1
Tall Man Lettering List REPORT DECEMBER 2013 1 TALL MAN LETTERING LIST REPORT WWW.HQSC.GOVT.NZ Published in December 2013 by the Health Quality & Safety Commission. This document is available on the Health Quality & Safety Commission website, www.hqsc.govt.nz ISBN: 978-0-478-38555-7 (online) Citation: Health Quality & Safety Commission. 2013. Tall Man Lettering List Report. Wellington: Health Quality & Safety Commission. Crown copyright ©. This copyright work is licensed under the Creative Commons Attribution-No Derivative Works 3.0 New Zealand licence. In essence, you are free to copy and distribute the work (including other media and formats), as long as you attribute the work to the Health Quality & Safety Commission. The work must not be adapted and other licence terms must be abided. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nd/3.0/nz/ Copyright enquiries If you are in doubt as to whether a proposed use is covered by this licence, please contact: National Medication Safety Programme Team Health Quality & Safety Commission PO Box 25496 Wellington 6146 ACKNOWLEDGEMENTS The Health Quality & Safety Commission acknowledges the following for their assistance in producing the New Zealand Tall Man lettering list: • The Australian Commission on Safety and Quality in Health Care for advice and support in allowing its original work to be either reproduced in whole or altered in part for New Zealand as per its copyright1 • The Medication Safety and Quality Program of Clinical Excellence Commission, New South