Selective Inhibition of Prostaglandin D2 Biosynthesis in Human Mast Cells to Overcome Need for Multiple Receptor Antagonists: Biochemical Consequences
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of the Structure and Function of a Timp-1/Cd63 Complex and Its Relationship to an Mt1-Mmp/Cd63 Complex
Wayne State University Wayne State University Dissertations 1-1-2013 Analysis Of The trS ucture And Function Of A Timp-1/cd63 Complex And Its Relationship To An Mt1-Mmp/cd63 Complex Richard B. Warner Wayne State University, Follow this and additional works at: http://digitalcommons.wayne.edu/oa_dissertations Part of the Oncology Commons Recommended Citation Warner, Richard B., "Analysis Of The trS ucture And Function Of A Timp-1/cd63 Complex And Its Relationship To An Mt1-Mmp/ cd63 Complex" (2013). Wayne State University Dissertations. Paper 864. This Open Access Dissertation is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Dissertations by an authorized administrator of DigitalCommons@WayneState. ANALYSIS OF THE STRUCTURE AND FUNCTION OF A TIMP-1/CD63 COMPLEX AND ITS RELATIONSHIP TO AN MT1-MMP/CD63 COMPLEX by RICHARD BECKSTRAND WARNER DISSERTATION Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY 2013 MAJOR: CANCER BIOLOGY Approved by: Advisor Date © COPYRIGHT BY RICHARD BECKSTRAND WARNER 2013 All Rights Reserved DEDICATION This work is dedicated to my wonderful family. Beginning with my wife and three children, I want to acknowledge how important my family has been to me throughout this major undertaking in my life. Paramount in my life is my relationship to God and my spirituality, which I apply in all things that I do; my wife, Stephanie, has been an unfailing support in this aspect. It is a truly wonderful and humbling experience that I have had as a spouse and parent together with her. -

RI-Mediated Mast Cell Activation Ε of Fc Tetraspanin CD151 Is A
Tetraspanin CD151 Is a Negative Regulator of Fc εRI-Mediated Mast Cell Activation Hiam Abdala-Valencia, Paul J. Bryce, Robert P. Schleimer, Joshua B. Wechsler, Lucas F. Loffredo, Joan M. Cook-Mills, This information is current as Chia-Lin Hsu and Sergejs Berdnikovs of September 26, 2021. J Immunol 2015; 195:1377-1387; Prepublished online 1 July 2015; doi: 10.4049/jimmunol.1302874 http://www.jimmunol.org/content/195/4/1377 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2015/07/01/jimmunol.130287 Material 4.DCSupplemental http://www.jimmunol.org/ References This article cites 63 articles, 28 of which you can access for free at: http://www.jimmunol.org/content/195/4/1377.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 26, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2015 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Tetraspanin CD151 Is a Negative Regulator of Fc«RI-Mediated Mast Cell Activation Hiam Abdala-Valencia,* Paul J. -

Prostaglandin D2 and the Role of the DP1, DP2 and TP Receptors in the Control of Airway Reflex Events
ERJ Express. Published on October 16, 2014 as doi: 10.1183/09031936.00061614 ORIGINAL ARTICLE IN PRESS | CORRECTED PROOF Prostaglandin D2 and the role of the DP1, DP2 and TP receptors in the control of airway reflex events Sarah A. Maher1, Mark A. Birrell1,2, John J. Adcock1, Michael A. Wortley1, Eric D. Dubuis1, Sara J. Bonvini1, Megan S. Grace1 and Maria G. Belvisi1,2 Affiliations: 1Respiratory Pharmacology, National Heart and Lung Institute, Faculty of Medicine, Imperial College London, London, UK. 2MRC and Asthma UK Centre in Allergic Mechanisms of Asthma, Imperial College London, London, UK. Correspondence: Maria G. Belvisi, Imperial College London, Respiratory Pharmacology, NHLI, Sir Alexander Fleming Building, Exhibition Road, London, SW7 2AZ, UK. E-mail: [email protected] ABSTRACT Prostaglandin D2 (PGD2) causes cough and levels are increased in asthma suggesting that it may contribute to symptoms. Although the prostaglandin D2 receptor 2 (DP2) is a target for numerous drug discovery programmes little is known about the actions of PGD2 on sensory nerves and cough. We used human and guinea pig bioassays, in vivo electrophysiology and a guinea pig conscious cough model to assess the effect of prostaglandin D2 receptor (DP1), DP2 and thromboxane receptor antagonism on PGD2 responses. PGD2 caused cough in a conscious guinea pig model and an increase in calcium in airway jugular ganglia. Using pharmacology and receptor-deficient mice we showed that the DP1 receptor mediates sensory nerve activation in mouse, guinea pig and human vagal afferents. In vivo, PGD2 and a DP1 receptor agonist, but not a DP2 receptor agonist, activated single airway C-fibres. -

Altered Expression of CD63 and Exosomes in Scleroderma Dermal
Journal of Dermatological Science 84 (2016) 30–39 Contents lists available at ScienceDirect Journal of Dermatological Science journal homepage: www.jdsjournal.com Altered expression of CD63 and exosomes in scleroderma dermal fibroblasts Kayo Nakamura, Masatoshi Jinnin*, Miho Harada, Hideo Kudo, Wakana Nakayama, Kuniko Inoue, Aki Ogata, Ikko Kajihara, Satoshi Fukushima, Hironobu Ihn Department of Dermatology and Plastic Surgery, Faculty of Life Sciences, Kumamoto University, 1-1-1 Honjo, Kumamoto 860-8556, Japan A R T I C L E I N F O A B S T R A C T Article history: Background: Exosomes are small vesicles shed from various cells. They contain proteins, lipids, and Received 6 January 2016 nucleic acids, and are regarded as a tool of cell-cell communication. Received in revised form 13 June 2016 Objectives: To reveal the putative role of exosomes in systemic sclerosis (SSc), and to elucidate the effect of Accepted 29 June 2016 exosomes on wound healing. Methods: The expression of common markers for exosomes (CD63, CD9, and CD81) and type I collagen Keywords: were examined with real-time PCR, immunohistochemical analysis, ELISA, immunoblotting, and flow Exosomes cytometry. The effect of serum-derived exosomes on wound healing was tested on full-thickness wounds CD63 in the mid-dorsal skin of BALB/c mice. Systemic sclerosis Results: The expression levels of CD63 as well as CD9 and CD81 tended to be increased in SSc dermal fibroblasts compared to normal fibroblasts. Increased exosomes in a cultured media of SSc fibroblasts stimulated the expression levels of type I collagen in normal fibroblasts. As the mechanism, collagen- related microRNA levels in SSc fibroblast-derived exosomes were dysregulated, indicating that both the amount and the content of exosomes were altered in SSc. -

A Shared Pathway of Exosome Biogenesis Operates at Plasma And
bioRxiv preprint doi: https://doi.org/10.1101/545228; this version posted February 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. A shared pathway of exosome biogenesis operates at plasma and endosome membranes Francis K. Fordjour1, George G. Daaboul2, and Stephen J. Gould1* 1Department of Biological Chemistry Johns Hopkins University Baltimore, MD USA 2Nanoview Biosciences Boston, MA USA Corresponding author: Stephen J. Gould, Ph.D. Department of Biological Chemistry Johns Hopkins University Baltimore, MD USA Email: [email protected] Tel (01) 443 847 9918 1 bioRxiv preprint doi: https://doi.org/10.1101/545228; this version posted February 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Summary: This study of exosome cargo protein budding reveals that cells use a common pathway for budding exosomes from plasma and endosome membranes, providing a new mechanistic explanation for exosome heterogeneity and a rational roadmap for exosome engineering. Keywords: Protein budding, tetraspanin, endosome, plasma membrane, extracellular vesicle, CD9, CD63, CD81, SPIR, interferometry Abbreviations: EV, extracellular vesicles; IB, immunoblot; IFM, immunofluorescence microscopy; IPMC, intracellular plasma membrane-connected compartment; MVB, multivesicular body; SPIR, single-particle interferometric reflectance; SPIRI, single-particle interferometric reflectance imaging 2 bioRxiv preprint doi: https://doi.org/10.1101/545228; this version posted February 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. -

Expression of the Tetraspanins CD9, CD37, CD63, and CD151 in Merkel Cell Carcinoma: Strong Evidence for a Posttranscriptional Fine-Tuning of CD9 Gene Expression
Modern Pathology (2010) 23, 751–762 & 2010 USCAP, Inc. All rights reserved 0893-3952/10 $32.00 751 Expression of the tetraspanins CD9, CD37, CD63, and CD151 in Merkel cell carcinoma: strong evidence for a posttranscriptional fine-tuning of CD9 gene expression Markus Woegerbauer1, Dietmar Thurnher1, Roland Houben2, Johannes Pammer3, Philipp Kloimstein1, Gregor Heiduschka1, Peter Petzelbauer4 and Boban M Erovic1 1Department of Otorhinolaryngology, Head and Neck Surgery, Medical University of Vienna, Vienna, Austria; 2Department of Dermatology, Medical University of Wuerzburg, Germany; 3Department of Clinical Pathology, Medical University of Vienna, Vienna, Austria and 4Department of Dermatology, Medical University of Vienna, Vienna, Austria Tetraspanins including CD9, CD37, CD63, and CD151 are linked to cellular adhesion, cell differentiation, migration, carcinogenesis, and tumor progression. The aim of the study was to detect, quantify, and evaluate the prognostic value of these tetraspanins in Merkel cell carcinoma and to study the regulation of CD9 mRNA expression in Merkel cell carcinoma cell lines in detail. Immunohistochemical staining of 28 Merkel cell carcinoma specimens from 25 patients showed a significant correlation of CD9 (P ¼ 0.03) and CD151 (P ¼ 0.043) expression to overall survival. CD9 and CD63 expression correlated significantly to patients’ disease-free interval (P ¼ 0.017 and P ¼ 0.058). Of primary Merkel cell carcinoma tumors, 42% were CD9 positive in contrast to only 21% of the subcutaneous in-transit metastases. Characterization of the 50 untranslated region (UTR) of the CD9 mRNA from two cultured Merkel cell carcinoma cell lines revealed the presence of two major RNA species differing only in the length of their 50 termini (183 versus 102 nucleotides). -

4 Supplementary File
Supplemental Material for High-throughput screening discovers anti-fibrotic properties of Haloperidol by hindering myofibroblast activation Michael Rehman1, Simone Vodret1, Luca Braga2, Corrado Guarnaccia3, Fulvio Celsi4, Giulia Rossetti5, Valentina Martinelli2, Tiziana Battini1, Carlin Long2, Kristina Vukusic1, Tea Kocijan1, Chiara Collesi2,6, Nadja Ring1, Natasa Skoko3, Mauro Giacca2,6, Giannino Del Sal7,8, Marco Confalonieri6, Marcello Raspa9, Alessandro Marcello10, Michael P. Myers11, Sergio Crovella3, Paolo Carloni5, Serena Zacchigna1,6 1Cardiovascular Biology, 2Molecular Medicine, 3Biotechnology Development, 10Molecular Virology, and 11Protein Networks Laboratories, International Centre for Genetic Engineering and Biotechnology (ICGEB), Padriciano, 34149, Trieste, Italy 4Institute for Maternal and Child Health, IRCCS "Burlo Garofolo", Trieste, Italy 5Computational Biomedicine Section, Institute of Advanced Simulation IAS-5 and Institute of Neuroscience and Medicine INM-9, Forschungszentrum Jülich GmbH, 52425, Jülich, Germany 6Department of Medical, Surgical and Health Sciences, University of Trieste, 34149 Trieste, Italy 7National Laboratory CIB, Area Science Park Padriciano, Trieste, 34149, Italy 8Department of Life Sciences, University of Trieste, Trieste, 34127, Italy 9Consiglio Nazionale delle Ricerche (IBCN), CNR-Campus International Development (EMMA- INFRAFRONTIER-IMPC), Rome, Italy This PDF file includes: Supplementary Methods Supplementary References Supplementary Figures with legends 1 – 18 Supplementary Tables with legends 1 – 5 Supplementary Movie legends 1, 2 Supplementary Methods Cell culture Primary murine fibroblasts were isolated from skin, lung, kidney and hearts of adult CD1, C57BL/6 or aSMA-RFP/COLL-EGFP mice (1) by mechanical and enzymatic tissue digestion. Briefly, tissue was chopped in small chunks that were digested using a mixture of enzymes (Miltenyi Biotec, 130- 098-305) for 1 hour at 37°C with mechanical dissociation followed by filtration through a 70 µm cell strainer and centrifugation. -
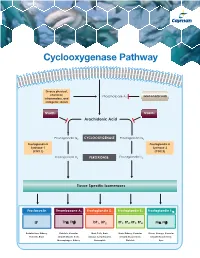
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

Extracellular Vesicle Human CD9/CD63/CD81 Antibody Panel
Extracellular Vesicle Human CD9/CD63/CD81 Antibody Panel Antibody panel for the detection of extracellular vesicles using CD9, CD63, and CD81 markers Catalog #100-0211 1 Kit Product Description The Extracellular Vesicle Human CD9/CD63/CD81 Antibody Panel is suitable for the detection of extracellular vesicles (EVs) derived from human cells. It comprises three primary antibodies that are immunoreactive toward human CD9, CD63, and CD81; these are proteins that are typically expressed on EVs and widely used as markers to analyze and isolate these cell-derived particles. CD9, CD63, and CD81 belong to the tetraspanin family of membrane proteins, which possess four transmembrane domains and interact with diverse proteins on the cell surface to form multimolecular networks termed tetraspanin-enriched microdomains. CD9, CD63, and CD81 proteins are expressed on the surface of many cells, including B cells, T cells, NK cells, monocytes, dendritic cells, thymocytes, endothelial cells, and fibroblasts, and are involved in modulating a variety of cellular processes including cell activation, adhesion, differentiation, and tumor invasion. The antibodies provided in this panel have been reported for use in analyzing primary cells, cell lines, and EVs by ELISA, flow cytometry, immunocytochemistry, immunoprecipitation, and Western blotting. They have been reported to cross-react with their cognate antigens in non-human primates, including baboons and rhesus and cynomolgus macaques. Product Information The following products comprise the Extracellular -

Aberrant Expression of Tetraspanin Molecules in B-Cell Chronic Lymphoproliferative Disorders and Its Correlation with Normal B-Cell Maturation
Leukemia (2005) 19, 1376–1383 & 2005 Nature Publishing Group All rights reserved 0887-6924/05 $30.00 www.nature.com/leu Aberrant expression of tetraspanin molecules in B-cell chronic lymphoproliferative disorders and its correlation with normal B-cell maturation S Barrena1,2, J Almeida1,2, M Yunta1,ALo´pez1,2, N Ferna´ndez-Mosteirı´n3, M Giralt3, M Romero4, L Perdiguer5, M Delgado1, A Orfao1,2 and PA Lazo1 1Instituto de Biologı´a Molecular y Celular del Ca´ncer, Centro de Investigacio´n del Ca´ncer, Consejo Superior de Investigaciones Cientı´ficas-Universidad de Salamanca, Salamanca, Spain; 2Servicio de Citometrı´a, Universidad de Salamanca and Hospital Universitario de Salamanca, Salamanca, Spain; 3Servicio de Hematologı´a, Hospital Universitario Miguel Servet, Zaragoza, Spain; 4Hematologı´a-hemoterapia, Hospital Universitario Rı´o Hortega, Valladolid, Spain; and 5Servicio de Hematologı´a, Hospital de Alcan˜iz, Teruel, Spain Tetraspanin proteins form signaling complexes between them On the cell surface, tetraspanin antigens are present either as and with other membrane proteins and modulate cell adhesion free molecules or through interaction with other proteins.25,26 and migration properties. The surface expression of several tetraspanin antigens (CD9, CD37, CD53, CD63, and CD81), and These interacting proteins include other tetraspanins, integri- F 22,27–30F their interacting proteins (CD19, CD21, and HLA-DR) were ns particularly those with the b1 subunit HLA class II 31–33 34,35 analyzed during normal B-cell maturation and compared to a moleculesFeg HLA DR -, CD19, the T-cell recep- group of 67 B-cell neoplasias. Three patterns of tetraspanin tor36,37 and several other members of the immunoglobulin expression were identified in normal B cells. -

Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Cr
Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Crizotinib (PF-02341066) 1 4 55 Docetaxel 1 5 98 Anastrozole 1 6 25 Cladribine 1 7 23 Methotrexate 1 8 -187 Letrozole 1 9 65 Entecavir Hydrate 1 10 48 Roxadustat (FG-4592) 1 11 19 Imatinib Mesylate (STI571) 1 12 0 Sunitinib Malate 1 13 34 Vismodegib (GDC-0449) 1 14 64 Paclitaxel 1 15 89 Aprepitant 1 16 94 Decitabine 1 17 -79 Bendamustine HCl 1 18 19 Temozolomide 1 19 -111 Nepafenac 1 20 24 Nintedanib (BIBF 1120) 1 21 -43 Lapatinib (GW-572016) Ditosylate 1 22 88 Temsirolimus (CCI-779, NSC 683864) 1 23 96 Belinostat (PXD101) 1 24 46 Capecitabine 1 25 19 Bicalutamide 1 26 83 Dutasteride 1 27 68 Epirubicin HCl 1 28 -59 Tamoxifen 1 29 30 Rufinamide 1 30 96 Afatinib (BIBW2992) 1 31 -54 Lenalidomide (CC-5013) 1 32 19 Vorinostat (SAHA, MK0683) 1 33 38 Rucaparib (AG-014699,PF-01367338) phosphate1 34 14 Lenvatinib (E7080) 1 35 80 Fulvestrant 1 36 76 Melatonin 1 37 15 Etoposide 1 38 -69 Vincristine sulfate 1 39 61 Posaconazole 1 40 97 Bortezomib (PS-341) 1 41 71 Panobinostat (LBH589) 1 42 41 Entinostat (MS-275) 1 43 26 Cabozantinib (XL184, BMS-907351) 1 44 79 Valproic acid sodium salt (Sodium valproate) 1 45 7 Raltitrexed 1 46 39 Bisoprolol fumarate 1 47 -23 Raloxifene HCl 1 48 97 Agomelatine 1 49 35 Prasugrel 1 50 -24 Bosutinib (SKI-606) 1 51 85 Nilotinib (AMN-107) 1 52 99 Enzastaurin (LY317615) 1 53 -12 Everolimus (RAD001) 1 54 94 Regorafenib (BAY 73-4506) 1 55 24 Thalidomide 1 56 40 Tivozanib (AV-951) 1 57 86 Fludarabine -

Effect of TTC-909 on Cerebral Infarction Following Permanent Occlusion of the Middle Cerebral Artery in Stroke Prone Spontaneously Hypertensive Rats
J Pharmacol Sci 91, 305 – 312 (2003) Journal of Pharmacological Sciences ©2003 The Japanese Pharmacological Society Full Paper Effect of TTC-909 on Cerebral Infarction Following Permanent Occlusion of the Middle Cerebral Artery in Stroke Prone Spontaneously Hypertensive Rats Yasuko Karasawa1, Hiroko Komiyama1, Shigeru Yoshida1, Noriko Hino1, Yasuhiro Katsuura2, Shiro Nakaike1 and Hiroaki Araki3,* 1Pharmacology Laboratory, Research Center, Taisho Pharmaceutical Co., Ltd., Yoshino-cho 1-403, Saitama 330-8530, Japan 2Pharmacological Research Department, Teijin Ltd., 4-3-2 Asahigaoka, Hino, Tokyo 191-8512, Japan 3Department of Hospital Pharmacy, Ehime University School of Medicine, Shiktsukawa, Shigenobu-cho, Onsen-gun, Ehime 791-0295, Japan Received November 14, 2002; Accepted January 30, 2003 Abstract. We investigated the effect of TTC-909, a drug preparation of the stable prostaglandin I2 analogue clinprost (isocarbacyclin methylester; methyl 5-{(1S,5S,6R,7R)-7- hydroxy-6-[(E)-(S)-3-hydroxy-1-octenyl] bicyclo[3.3.0]oct-2-en-3-yl} pentanoate) incorporated into lipid microspheres, on cerebral infarction 7 days after permanent occlusion of the middle cerebral artery (MCA) in stroke prone spontaneously hypertensive rats (SHRSP). Under the anesthesia, the MCA was permanently occluded above the rhinal fissure. In schedule 1, vehicle or TTC-909 was injected i.v. once daily over 7 days starting immediately after MCA occlusion. In schedule 2, vehicle or TTC-909 was infused for 3 h starting immediately after MCA occlusion. In schedule 3, vehicle or TTC-909 was infused for 3 h starting immediately after MCA occlusion followed by bolus injection once daily over 6 days. Seven days later, the infarct volume was estimated following hematoxylin and eosin staining.