Edwin M. Mcmillan
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

February 2020 Newsletter Manhattan Project NHP Oak Ridge
National Park Service February 2020 Department of the Interior Manhattan Project National Historical Park Oak Ridge, Tennessee Manhattan Project History in February Visit Inside the Oak Ridge Turnpike Gatehouse on Sat- Philip Abelson began working on uranium enrichment urday, February 8 at 3:30 pm (ET). Rangers will host the using liquid thermal diffusion in February of 1941. How- popular “Secrecy, Security, and Spies” program at the Turn- ever construction of his thermal diffusion plant in Phila- pike Gatehouse. The program will begin at 3:30 pm (ET) giv- delphia did not begin until January 1944. This same pro- ing visitors some insight to what life was like in Oak Ridge cess would be used later at the S-50 plant in Oak Ridge, during the Manhattan Project with the need for secrecy, all TN. of the security that surrounded the city, and the threat of Glenn Seaborg and his research group discovered Pluto- spies. The Turnpike Gatehouse is located at 2900 Oak Ridge nium while working at the University of California, Berke- Turnpike, Oak Ridge, TN 37830. ley on February 24, 1941. Construction of the -Y 12 Ura- Join a Park Ranger for a nium enrichment facilities and “Manhattan Project in Pop the X-10 Graphite Reactor be- Culture” on Saturday, Febru- gan in February 1943. Stone ary 22 at 3 pm (ET) at the Ameri- and Webster Engineering was can Museum of Science & Energy, in charge of Y-12 construction 115 East Main Street, Oak Ridge. while DuPont handled X-10. We will lead a discussion of the One year after construction Y- impact the Project had on popu- 12 sent 200 grams of uranium- lar culture in movies, music and 235 to Los Alamos, NM in Feb- television. -

Science Loses Some Friends Francis Crick, Thomas Gold, and Philip Abelson
Monthly Planet October 2004 Science Loses Some Friends Francis Crick, Thomas Gold, and Philip Abelson By Iain Murray he scientifi c world lost three impor- Throughout his career, Abelson used which suggests that the universe and Ttant fi gures in recent weeks, as scientifi c principles to determine genu- the laws of physics have always existed Francis Crick, Thomas Gold, and Philip ine new developments from hype and in the same, steady state. This has since Abelson have all passed away. In their publicity stunts (he was famously dis- been supplanted as the dominant cos- careers, each demonstrated the best that missive of the scientifi c value of the race mological paradigm by the Big Bang science has to offer humanity. Their loss to the moon). In that, he should prove a theory. illustrates how much worse off the state role model for true scientists. After working at the Royal Greenwich of science is today than during their Observatory, Gold moved to the United glory years. Thomas Gold States to become Professor of Astron- Astronomer Thomas Gold had an omy at Harvard. From there he moved Philip Abelson equally distinguished career, in fi elds as to Cornell, where he demonstrated that Philip Abelson was a scientist of truly diverse as engineering, physiology, and the newly discovered “pulsar” phenom- broad talents. One of America’s fi rst cosmology; and he was never afraid of enon must contain a rotating neutron nuclear physicists, he discovered the being called a maverick. A fellow of both star (a star more massive than the sun element Neptunium and designed the the Royal Society and the National Acad- but just 10 km in diameter). -

Ija, LLP Alan D
20-12212-mew Doc 1196 Filed 05/07/21 Entered 05/07/21 21:22:15 Main Document Pg 1 of 74 UNITED STATES BANKRUPTCY COURT SOUTHERN DISTRICT OF NEW YORK -----------------------------------------------------------------------x : In re: : Chapter 11 : GARRETT MOTION INC., et al.,1 : Case No. 20-12212 (MEW) : Debtors. : (Jointly Administered) : -----------------------------------------------------------------------x CERTIFICATE OF SERVICE I, Rossmery Martinez, depose and say that I am employed by Kurtzman Carson Consultants LLC (KCC), the claims and noticing agent for the Debtors in the above-captioned case. On May 4, 2021, at my direction and under my supervision, employees of KCC caused to be served the following document via Electronic Mail upon the service list attached hereto as Exhibit A; and via First Class Mail upon the service list attached hereto as Exhibit B: Notice of June Omnibus Hearing Date [Docket No. 1192] Furthermore, on May 4, 2021, at my direction and under my supervision, employees of KCC caused to be served the following document via Electronic Mail upon the service list attached hereto as Exhibit A; and via First Class Mail upon the service lists attached hereto as Exhibit B and Exhibit C: (Continued on Next Page) 1 The last four digits of Garrett Motion Inc.’s tax identification number are 3189. Due to the large number of debtor entities in these Chapter 11 Cases, which are being jointly administered, a complete list of the Debtors and the last four digits of their federal tax identification numbers is not provided herein. A complete list of such information may be obtained on the website of the Debtors’ claims and noticing agent at http://www.kccllc.net/garrettmotion. -
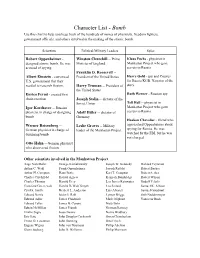
Character List
Character List - Bomb Use this chart to help you keep track of the hundreds of names of physicists, freedom fighters, government officials, and others involved in the making of the atomic bomb. Scientists Political/Military Leaders Spies Robert Oppenheimer - Winston Churchill -- Prime Klaus Fuchs - physicist in designed atomic bomb. He was Minister of England Manhattan Project who gave accused of spying. secrets to Russia Franklin D. Roosevelt -- Albert Einstein - convinced President of the United States Harry Gold - spy and Courier U.S. government that they for Russia KGB. Narrator of the needed to research fission. Harry Truman -- President of story the United States Enrico Fermi - created first Ruth Werner - Russian spy chain reaction Joseph Stalin -- dictator of the Tell Hall -- physicist in Soviet Union Igor Korchatov -- Russian Manhattan Project who gave physicist in charge of designing Adolf Hitler -- dictator of secrets to Russia bomb Germany Haakon Chevalier - friend who Werner Reisenberg -- Leslie Groves -- Military approached Oppenheimer about German physicist in charge of leader of the Manhattan Project spying for Russia. He was designing bomb watched by the FBI, but he was not charged. Otto Hahn -- German physicist who discovered fission Other scientists involved in the Manhattan Project: Aage Niels Bohr George Kistiakowsky Joseph W. Kennedy Richard Feynman Arthur C. Wahl Frank Oppenheimer Joseph Rotblat Robert Bacher Arthur H. Compton Hans Bethe Karl T. Compton Robert Serber Charles Critchfield Harold Agnew Kenneth Bainbridge Robert Wilson Charles Thomas Harold Urey Leo James Rainwater Rudolf Pelerls Crawford Greenewalt Harold DeWolf Smyth Leo Szilard Samuel K. Allison Cyril S. Smith Herbert L. Anderson Luis Alvarez Samuel Goudsmit Edward Norris Isidor I. -

Feb 05 Nucleus Corrected
DED UN 18 O 98 F yyyy N yyyy Y O T R E I T H C E N O yyyy A E S S S L T A E A C R C I yyyyN S M S E E H C C T N IO A February 2005 Vol. LXXXIII, No. 6 yyyyC N • AMERI Monthly Meeting Prof. Shana Kelley on Nanotechnological and Biomolecules Arno Heyn Dedicated NESACS Member Organohalogens Gordon Gribble Writes Philip Abelson A National Figure in Science Call for Papers 20052005 ❖ Deadline – April 15, 2005 ❖ EASTERN November 14-17, 2005 ANALYTICAL Garden State Exhibit Center Somerset, NJ SYMPOSIUM he Eastern Analytical Symposium is the second largest meeting in the United States dedicated to the needs Innovation through Education of analytical chemists and those in the allied sciences. Please help us to make the 2005 EAS the best ever–be a part of the program by contributing your own papers for inclusion in the oral or poster sessions. To submit a contributed presentation for the 2005 EAS Technical Program, you should go to our web site, www.eas.org, after March 1, and follow the instructions for preliminary abstract submission. Invited speakers should not submit preliminary abstracts to EAS, although your session organizer may request one for his/her use. All preliminary abstracts must be submitted elec- tronically via the EAS web site at www.eas.org. The abstract submis- sion deadline is April 15, 2005. No faxed, e-mailed, or mailed prelimi- nary abstracts will be accepted. Please carefully review the following information: 1. -

National Park Service Department of the Interior
National Park Service May 2017 Department of the Interior Manhattan Project National Historical Park Oak Ridge, Tennessee May in MAPR History April Showers Brings May Flowers and Also Cabbage, British physicist James Chadwick would later be awarded the Tomatoes and Green Beans. We are hard at work in the Nobel Prize in Physics for his discovery of the neutron in May Victory Garden next to the Flat Top House with students 1932. from Willow Brook Elementary every Wednesday. Students Gorge Kistiakowsky and Vannevar Bush have been planting and caring for the garden discussed the possibility of producing U- and learning about war time rationing. 235 by using gaseous diffusion in May 1940. On May 27, 1940, the discovery of radioac- Learn about the Upcoming Total Solar tive element 93, neptunium, is announced Eclipse on Thursday, May 18. Join us for a in a report submitted by Edwin McMillan lecture featuring Chap Percival who will and Philip Abelson. The same day Leo Szil- speak on the upcoming August 21 solar ard received a manuscript from Louis eclipse. This free lecture will be held at the Turner arguing that the isotope of element American Museum of Science and Energy. A 94, still undiscovered plutonium, should reception will start at 5:30 p.m. (ET) and the be highly fissionable like uranium-235. lecture starts at 6 p.m. Glenn Seaborg was able to prove on May 3, 1941, that plutonium-239 was more fissionable than uranium-235 through his This Month’s AMSE Curatorial Collection research at the University of California, Highlight is Jack Beers’ Library Card from Berkeley. -

Edwin M. Mcmillan, a Biographical Sketch
LBL 35965 HQDFAN668 EDWIN M. MCMILLAN, A BIOGRAPHICAL SKETCH EDWARD J. LOFGREN, Associate Director Emeritus LAWRENCE BERKELEY LABORATORY BERKELEY, CALIFORNIA 94720 Presented at the International Symposium: The 50th Anniversary of the Phase Stability Principle Moscow/Dubna, Russia 12-15 July 1994 ABSTRACT. Edwin M. McMillan was one of the great scientists of the middle years of this century. He made notable contributions to nuclear, and particle physics, the chemis try of transuranic elements, and accelerator physics. * This work supported by the Director, Office of Energy Research, Office of Fusion Energy, U.S. Department of Energy under contract No. DE-AC03-76SF00098. ^-TWBUTION OF THIS DOCUMENT (S UNLIMITED Edwin McMillan was born on Septem tion Laboratory. He joined the Laboratory in ber 18,1907 at Redondo Beach, California. Soon 1934 and began a lifelong close association with after, the family moved to Pasadena, California Lawrence. where the father practised medicine for many He quickly established himself as a me years. The move to Pasadena was a very fortu ticulous and versatile experimenter in nuclear nate one because Caltech was nearby and even physics with an excellent grasp of theory. Dur in his early school years Ed could take advan ing this period he discovered *-0 with Stanley tage of the excellent public programs and lec Livingston and 10Be with Samuel Ruben. Per tures put on there by them. This contact with the haps his best experiment at that time was the world of science as Ed grew from boyhood to a demonstration of electron pair production by the young man was very important, for it nurtured absorption of gamma rays from fluorine bom the wide and lasting curiosity and interest that barded by protons from the cyclotron. -

An Atomic History Chapter 2
An Atomic History 0-3 8/11/02 7:31 AM Page 18 Chapter Two 19 THE FERMI-SZILARD PILE AND URANIUM RESEARCH The first government funding for nuclear research was allocated to purchase graphite and uranium oxide for the chain reaction experiments being organized by Fermi and World War II and the Manhattan Project Szilard at Columbia University in February 1940.2 This work, which began in New York 2 City, soon spread to Princeton, the University of Chicago, and research institutions in California.3 Even at this stage, the scientists knew that a chain reaction would need three major components in the right combination: fuel, moderator, and coolant. The fuel would contain the fissile material needed to support the fission process. The neutrons generated by the fission process had to be slowed by the moderator so that they could initiate addi- tional fission reactions. The heat that resulted from this process had to be removed by the coolant. Fermi’s initial research explored the possibility of a chain reaction with natural urani- The 1930s were a time of rapid progress in the development of nuclear physics. um. It was quickly determined that high-purity graphite served as the best neutron moder- Research accelerated in the early years of the Second World War, when new developments ator out of the materials then available.4 After extensive tests throughout 1940 and early were conceived and implemented in the midst of increasing wartime urgency. American 1941, Fermi and Szilard set up the first blocks of graphite at Columbia University in government interest in these developments was limited at first, but increased as the war September 1941. -

The Manhattan Project: Making the Atomic Bomb” Is a Short History of the Origins and Develop- Ment of the American Atomic Bomb Program During World War H
f.IOE/MA-0001 -08 ‘9g [ . J vb JMkirlJkhilgUimBA’mmml — .— Q RDlmm UNITED STATES DEPARTMENT OF ENERGY ,:.. .- ..-. .. -,.,,:. ,.<,.;<. ~-.~,.,.- -<.:,.:-,------—,.--,,p:---—;-.:-- ---:---—---- -..>------------.,._,.... ,/ ._ . ... ,. “ .. .;l, ..,:, ..... ..’, .’< . Copies of this publication are available while supply lasts from the OffIce of Scientific and Technical Information P.O. BOX 62 Oak Ridge, TN 37831 Attention: Information Services Telephone: (423) 576-8401 Also Available: The United States Department of Energy: A Summary History, 1977-1994 @ Printed with soy ink on recycled paper DO13MA-0001 a +~?y I I Tho PROJEOT UNITED STATES DEPARTMENT OF ENERGY F.G. Gosling History Division Executive Secretariat Management and Administration Department of Energy ]January 1999 edition . DISCLAIMER This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, make any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. I DISCLAIMER Portions of this document may be illegible in electronic image products. Images are produced from the best available original document. 1 Foreword The Department of Energy Organization Act of 1977 brought together for the first time in one department most of the Federal Government’s energy programs. -

Uranium Enrichment Processes Directed Self-Study Course! This Is the Fifth of Seven Modules Available in This Self-Study Course
MODULE 5.0: ELECTROMAGNETIC SEPARATION (CALUTRON) AND THERMAL DIFFUSION Introduction Welcome to Module 5.0 of the Uranium Enrichment Processes Directed Self-Study Course! This is the fifth of seven modules available in this self-study course. The purpose of this module is to assist the trainee in describing the general principles of the electromagnetic separation (calutron) and thermal diffusion technologies and general facility and component layouts, identifying the uses of the calutron and thermal diffusion processes in industry and the production amounts of enriched uranium, and identifying the hazards and safety concerns for each process, including major incidents. This self-study module is designed to assist you in accomplishing the learning objectives listed at the beginning of the module. There are eight sections in this module. The module has self-check questions to help you assess your understanding of the concepts presented in the module. Before you Begin It is recommended that you have access to the following materials: 9 Trainee Guide Complete the following prerequisite: 9 Module 1.0 Introduction to Uranium Enrichment 1. Review the learning objectives. 2. Read each section within the module in sequential order. How to Complete 3. Complete the self-check questions and activities within this This Module module. 4. Check off the tracking form as you complete the self-check questions and/or activity within the module. 5. Contact your administrator as prompted for a progress review meeting. 6. Contact your administrator as prompted for any additional materials and/or specific assignments. 7. Complete all assignments related to this module. If no other materials or assignments are given to you by your administrator, you have completed this module. -

History of the Seaborg Institute
HISTORY OF THE SEABORG INSTITUTE ARQ.2_09.cover.indd 1 6/22/09 9:24 AM ContentsContents History of the Seaborg Institute The Seaborg Institute for Transactinium Science Established to educate the next generation of nuclear scientists 1 ITS Advisory Council 5 Seaborg the scientist 7 Naming seaborgium 11 Branching out 13 IGCAR honors Seaborg’s contributions to actinide science 16 Seaborg Institute for Transactinium Science/Los Alamos National Laboratory Actinide Research Quarterly The Seaborg Institute for Transactinium Science Established to educate the next generation of nuclear scientists In the late 1970s and early 1980s, research opportunities in heavy element This article was contributed by science and engineering were seldom found in university settings, and the supply Darleane C. Hoffman, professor emerita, of professionals in those fields was diminishing to the detriment of national Graduate School, Department of goals. In response to these concerns, national studies were conducted and panels Chemistry, UC Berkeley, faculty senior and committees were formed to assess the status of training and education in the scientist, Lawrence Berkeley National nuclear sciences. The studies in particular addressed the future need for scientists Laboratory, and charter director, Seaborg trained in the areas of nuclear waste management and disposal, environmental Institute, Lawrence Livermore National remediation, nuclear fuel processing, and nuclear safety analysis. Laboratory; and Christopher Gatrousis, Unfortunately, with the exception of the American Chemical Society’s former associate director of Chemistry Division of Nuclear Chemistry and Technology summer schools for undergradu- and Materials Science, Lawrence ates in nuclear and radiochemistry established in 1984, the studies did not lead Livermore National Laboratory. -

Philip H. Abelson
Glossary on Kalinga Prize Laureates UNESCO – Kalinga Prize Winner – 1972 Philip H. Abelson World Renowned Scientist, Longtime Editor of ‘Science’ and Co-Discoverer of the Element Neptunium. [Born : April 27, 1913, Tacoma, WA, USA Died : August 1, 2004 at Suburban Hospital in Bethesda, Maryland, USA] Organic matter equivalent in quantity to the weight of the Earth has been created by living creatures since life originated on this planet. ... Philip H. Abelson 1957 “Man is the Product of billions of years of hard-won evolution”. We must notrisk permitting Zealots, however well-intentioned, to gamble with the future. .... Philip H. Abelson, 1966 The imaginative and original mind need not be overawed by the imposing body of Present knowledge or by the complex and costly paraphernalia which today surround much of scientific activity. The great shortage in science now is not opportunity, manpower, money or laboratory space. What is really needed is more of that healthy skepticism which generates the Key ideas - the liberating concept. ....Philip H. Abelson 1 Glossary on Kalinga Prize Laureates Philip H. Abelson Philip Hauge Abelson (April 27, 1913 - August 1, He was a key contributor to the Manhattan Project 2004) was an American physicist, editor of scientific during World War II. Although Abelson was not literature, and science writer. formally associated with the atom bomb project, the Liquid Thermal Diffusion isotope separation technique Philip Abelson was born in 1913 in Tacoma, that he invented was used in the S-50 plant in Oak Washington. He attended Washington State Ridge, Tennessee, and proved a critical step in University where he received degrees in Chemistry creating sufficient fuel for the weapon.