Nucleobindin-1/2-Encoded Insulinotropic and Enterotropic Peptides
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Studies on the Roles of Translationally Recoded Proteins from Cyclooxygenase-1 and Nucleobindin Genes in Autophagy Jonathan J
Brigham Young University BYU ScholarsArchive All Theses and Dissertations 2015-06-01 Studies on the Roles of Translationally Recoded Proteins from Cyclooxygenase-1 and Nucleobindin Genes in Autophagy Jonathan J. Lee Brigham Young University Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Chemistry Commons BYU ScholarsArchive Citation Lee, Jonathan J., "Studies on the Roles of Translationally Recoded Proteins from Cyclooxygenase-1 and Nucleobindin Genes in Autophagy" (2015). All Theses and Dissertations. 6538. https://scholarsarchive.byu.edu/etd/6538 This Dissertation is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in All Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Studies on the Roles of Translationally Recoded Proteins from Cyclooxygenase-1 and Nucleobindin Genes in Autophagy Jonathan J. Lee A dissertation submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Doctor of Philosophy Daniel L. Simmons, Chair Richard K. Watt Joshua L. Andersen Barry M. Willardson Jeffery Barrow Department of Chemistry and Biochemistry Brigham Young University June 2015 Copyright © 2015 Jonathan J. Lee All Rights Reserved ABSTRACT Studies on the Roles of Translationally Recoded Proteins from Cyclooxygenase-1 and Nucleobindin Genes in Autophagy Jonathan J. Lee Department of Chemistry and Biochemistry, BYU Doctor of Philosophy Advances in next-generation sequencing and ribosomal profiling methods highlight that the proteome is likely orders of magnitude larger than previously thought. This expansion potentially occurs through translational recoding, a process that results in the expression of multiple variations of a protein from a single messenger RNA. -

Original Article Nucleobindin 2 (NUCB2) in Renal Cell Carcinoma: a Novel Factor Associated with Tumor Development
Int J Clin Exp Med 2019;12(7):8686-8693 www.ijcem.com /ISSN:1940-5901/IJCEM0090807 Original Article Nucleobindin 2 (NUCB2) in renal cell carcinoma: a novel factor associated with tumor development Ziwei Wei*, Huan Xu*, Qiling Shi*, Long Li, Juan Zhou, Guopeng Yu, Bin Xu, Yushan Liu, Zhong Wang, Wenzhi Li Department of Urology, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, P.R. China. *Equal contributors. Received January 3, 2019; Accepted May 8, 2019; Epub July 15, 2019; Published July 30, 2019 Abstract: This study aimed to examine the expression of NUCB2 in renal cell carcinoma (RCC) tissues and detect its effect on the apoptosis and proliferation of RCC cells both in vivo and vitro. NUCB2 was detected with higher expres- sion in the RCC tissues compared with the adjacent noncancerous tissues by immunohistochemical analysis (P < 0.05). Moreover, the protein levels of NUCB2 was significantly associated with perinephric tissues invasion, cancer- ous thrombus and distant metastasis. NUCB2 knock-down inhibited cell proliferation by arresting the cell cycle at S phase and increased the cell apoptosis detected by CCK-8 and flow cytometry analysis. Finally, the tumor-bearing mice models were constructed through injection of 786-O cells transfected with lentivirus carrying shRNAs targeting NUCB2 or ACTB cDNA plasmids. As expected, the mice in the NUCB2-shRNA group demonstrated a reduced tumor volume and growth rate compared with that in negative control group. This study observed the up-regulated expres- sion of NUCB2 in the ccRCC tissue and 786-O cell line. -

Genome-Wide Analysis of Organ-Preferential Metastasis of Human Small Cell Lung Cancer in Mice
Vol. 1, 485–499, May 2003 Molecular Cancer Research 485 Genome-Wide Analysis of Organ-Preferential Metastasis of Human Small Cell Lung Cancer in Mice Soji Kakiuchi,1 Yataro Daigo,1 Tatsuhiko Tsunoda,2 Seiji Yano,3 Saburo Sone,3 and Yusuke Nakamura1 1Laboratory of Molecular Medicine, Human Genome Center, Institute of Medical Science, The University of Tokyo, Tokyo, Japan; 2Laboratory for Medical Informatics, SNP Research Center, Riken (Institute of Physical and Chemical Research), Tokyo, Japan; and 3Department of Internal Medicine and Molecular Therapeutics, The University of Tokushima School of Medicine, Tokushima, Japan Abstract Molecular interactions between cancer cells and their Although a number of molecules have been implicated in microenvironment(s) play important roles throughout the the process of cancer metastasis, the organ-selective multiple steps of metastasis (5). Blood flow and other nature of cancer cells is still poorly understood. To environmental factors influence the dissemination of cancer investigate this issue, we established a metastasis model cells to specific organs (6). However, the organ specificity of in mice with multiple organ dissemination by i.v. injection metastasis (i.e., some organs preferentially permit migration, of human small cell lung cancer (SBC-5) cells. We invasion, and growth of specific cancer cells, but others do not) analyzed gene-expression profiles of 25 metastatic is a crucial determinant of metastatic outcome, and proteins lesions from four organs (lung, liver, kidney, and bone) involved in the metastatic process are considered to be using a cDNA microarray representing 23,040 genes and promising therapeutic targets. extracted 435 genes that seemed to reflect the organ More than a century ago, Stephen Paget suggested that the specificity of the metastatic cells and the cross-talk distribution of metastases was not determined by chance, but between cancer cells and microenvironment. -

Deep Sequencing of the Zebrafish Transcriptome Response to Mycobacterium Infection
Transcriptome profiling of infectious diseases and cancer in zebrafish Ordas, A.K. Citation Ordas, A. K. (2010, June 29). Transcriptome profiling of infectious diseases and cancer in zebrafish. Retrieved from https://hdl.handle.net/1887/15734 Version: Not Applicable (or Unknown) License: Leiden University Non-exclusive license Downloaded from: https://hdl.handle.net/1887/15734 Note: To cite this publication please use the final published version (if applicable). Chapter 2 Deep sequencing of the zebrafish transcriptome response to mycobacterium infection This chapter is based on: Hegedus Z, Zakrzewska A, Agoston VC, Ordas A, Racz P, Mink M, Spaink HP, Meijer AH. Deep sequencing of the zebrafish transcriptome response to myco- bacterium infection. Mol Immunol. 2009 Sep;46(15):2918-30. Chapter 2 Abstract Novel high-throughput deep sequencing technology has dramatically changed the way that the functional complexity of transcriptomes can be studied. Here we report on the first use of this technology to gain insight into the wide range of transcrip- tional responses that are associated with an infectious disease process. Using Solexa/ Illumina’s digital gene expression (DGE) system, a tag-based transcriptome sequenc- ing method, we investigated mycobacterium-induced transcriptome changes in a model vertebrate species, the zebrafish. We obtained a sequencing depth of over 5 million tags per sample with strong correlation between replicates. Tag mapping indicated that healthy and infected adult zebrafish express over 70% of all genes represented in transcript databases. Comparison of the data with a previous mul- ti-platform microarray analysis showed that both types of technologies identified regulation of similar functional groups of genes. -

Genetic Analyses Uncover Pleiotropic Compensatory Roles for Drosophila Nucleobindin-1 in Inositol Trisphosphate- Mediated Intracellular Calcium Homeostasis
Genome Genetic analyses uncover pleiotropic compensatory roles for Drosophila Nucleobindin-1 in inositol trisphosphate- mediated intracellular calcium homeostasis Journal: Genome Manuscript ID gen-2019-0113.R2 Manuscript Type: Article Date Submitted by the 11-Sep-2019 Author: Complete List of Authors: Balasubramanian, Vidhya; Indian Institute of Technology Madras, Department of Biotechnology SRINIVASAN, BHARATH; Indian Institute of Technology Madras, DepartmentDraft of Biotechnology Intracellular calcium homeostasis, Golgi, IP<sub>3</sub> receptor, Keyword: <i>Drosophila melanogaster</i>, Nucleobindin-1 Is the invited manuscript for consideration in a Special Not applicable (regular submission) Issue? : https://mc06.manuscriptcentral.com/genome-pubs Page 1 of 86 Genome Page 1 of 49 1 Title Page 2 Genetic analyses uncover pleiotropic compensatory roles for Drosophila 3 Nucleobindin-1 in inositol trisphosphate-mediated intracellular calcium 4 homeostasis 5 6 Authors: Vidhya Balasubramanian1 and Bharath Srinivasan1* 7 1Department of Biotechnology, Bhupat and Jyoti Mehta School of Biosciences, Indian Institute 8 of Technology-Madras, Chennai, 600036, India. 9 Telephone: +9181222 48706 10 11 Email: [email protected] 12 *Corresponding Author 13 Draft 14 15 16 17 18 19 20 21 22 23 24 25 26 https://mc06.manuscriptcentral.com/genome-pubs Genome Page 2 of 86 Page 2 of 49 27 Abstract: 28 Nucleobindin-1 is an EF-hand calcium-binding protein with a distinctive profile, predominantly 29 localized to the Golgi in insect and wide-ranging vertebrate cell types, alike. Its putative 30 involvements in intracellular calcium (Ca2+) homeostasis have however never been 31 phenotypically characterized in any model organism. We have analyzed an adult-viable mutant 32 that completely disrupts the G protein α-subunit Binding and Activating (GBA) motif of 33 Drosophila Nucleobindin-1 (dmNUCB1). -

Comparison of Human Chromosome 19Q13 and Syntenic Region On
European Journal of Human Genetics (2010) 18, 442–447 & 2010 Macmillan Publishers Limited All rights reserved 1018-4813/10 $32.00 www.nature.com/ejhg ARTICLE Comparison of human chromosome 19q13 and syntenic region on mouse chromosome 7 reveals absence, in man, of 11.6 Mb containing four mouse calcium-sensing receptor-related sequences: relevance to familial benign hypocalciuric hypercalcaemia type 3 Fadil M Hannan1, M Andrew Nesbit1, Jeremy JO Turner1,2, Joanna M Stacey1, Luisella Cianferotti3, Paul T Christie1, Arthur D Conigrave4, Michael P Whyte5,6 and Rajesh V Thakker*,1 Familial benign hypocalciuric hypercalcaemia (FBHH) is a genetically heterogeneous disorder that consists of three designated types, FBHH1, FBHH2 and FBHH3, whose chromosomal locations are 3q21.1, 19p and 19q13, respectively. FBHH1 is caused by mutations of a calcium-sensing receptor (CaSR), but the abnormalities underlying FBHH2 and FBHH3 are unknown. FBHH3, also referred to as the Oklahoma variant (FBHHOk), has been mapped to a 12cM interval, flanked by D19S908 and D19S866. To refine the location of FBHH3, we pursued linkage studies using 24 polymorphic loci. Our results establish a linkage between FBHH3 and 17 of these loci, and indicate that FBHH3 is located in a 4.1 Mb region flanked centromerically by D19S112 and telomerically by rs245111, which in the syntenic region on mouse chromosome 7 contains four Casr-related sequences (Gprc2a-rss). However, human homologues of these Gprc2a-rss were not found and a comparative analysis of the 22.0 Mb human and 39.3 Mb mouse syntenic regions showed evolutionary conservation of two segments that were inverted with loss from the human genome of 11.6 Mb that contained the four Gprc2a-rss. -

Universidad Autónoma De Madrid
Universidad Autónoma de Madrid Departamento de Bioquímica Identification of the substrates of the protease MT1-MMP in TNFα- stimulated endothelial cells by quantitative proteomics. Analysis of their potential use as biomarkers in inflammatory bowel disease. Agnieszka A. Koziol Tesis doctoral Madrid, 2013 2 Departamento de Bioquímica Facultad de Medicina Universidad Autónoma de Madrid Identification of the substrates of the protease MT1-MMP in TNFα- stimulated endothelial cells by quantitative proteomics. Analysis of their potential use as biomarkers in inflammatory bowel disease. Memoria presentada por Agnieszka A. Koziol licenciada en Ciencias Biológicas para optar al grado de Doctor. Directora: Dra Alicia García Arroyo Centro Nacional de Investigaciones Cardiovasculares (CNIC) Madrid, 2013 3 4 ACKNOWLEDGEMENTS - ACKNOWLEDGEMENTS - First and foremost I would like to express my sincere gratitude to my supervisor Dr. Alicia García Arroyo for giving me the great opportunity to study under her direction. Her encouragement, guidance and support from the beginning to the end, enabled me to develop and understand the subject. Her feedback to this manuscript, critical reading and corrections was inappreciable to finish this dissertation. Next, I would like to express my gratitude to those who helped me substantially along the way. To all members of MMPs’ lab, for their interest in the project, teaching me during al these years and valuable contribution at the seminary discussion. Without your help it would have not been possible to do some of those experiments. Thank you all for creating a nice atmosphere at work and for being so good friends in private. I would like to also thank all of the collaborators and technical units for their support and professionalism. -

Activation of the Endogenous Renin-Angiotensin- Aldosterone System Or Aldosterone Administration Increases Urinary Exosomal Sodium Channel Excretion
CLINICAL RESEARCH www.jasn.org Activation of the Endogenous Renin-Angiotensin- Aldosterone System or Aldosterone Administration Increases Urinary Exosomal Sodium Channel Excretion † † Ying Qi,* Xiaojing Wang, Kristie L. Rose,* W. Hayes MacDonald,* Bing Zhang, ‡ | Kevin L. Schey,* and James M. Luther § Departments of *Biochemistry, †Bioinformatics, ‡Division of Clinical Pharmacology, Department of Medicine, §Division of Nephrology, Department of Medicine, and |Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, Tennessee ABSTRACT Urinary exosomes secreted by multiple cell types in the kidney may participate in intercellular signaling and provide an enriched source of kidney-specific proteins for biomarker discovery. Factors that alter the exosomal protein content remain unknown. To determine whether endogenous and exogenous hormones modify urinary exosomal protein content, we analyzed samples from 14 mildly hypertensive patients in a crossover study during a high-sodium (HS, 160 mmol/d) diet and low-sodium (LS, 20 mmol/d) diet to activate the endogenous renin-angiotensin-aldosterone system. We further analyzed selected exosomal protein content in a separate cohort of healthy persons receiving intravenous aldosterone (0.7 mg/kg per hour for 10 hours) versus vehicle infusion. The LS diet increased plasma renin activity and aldosterone concentration, whereas aldosterone infusion increased only aldosterone concentration. Protein analysis of paired urine exosome samples by liquid chromatography-tandem mass spectrometry–based multidimen- sional protein identification technology detected 2775 unique proteins, of which 316 exhibited signifi- cantly altered abundance during LS diet. Sodium chloride cotransporter (NCC) and a-andg-epithelial sodium channel (ENaC) subunits from the discovery set were verified using targeted multiple reaction monitoring mass spectrometry quantified with isotope-labeled peptide standards. -

Quantitative Label-Free Proteomic Analysis of Milk Fat Globule Membrane in Donkey and Human Milk
ORIGINAL RESEARCH published: 22 June 2021 doi: 10.3389/fnut.2021.670099 Quantitative Label-Free Proteomic Analysis of Milk Fat Globule Membrane in Donkey and Human Milk Xinhao Zhang 1,2†, Bo Jiang 3†, Chuanliang Ji 2†, Haijing Li 2, Li Yang 2, Guimiao Jiang 2, Yantao Wang 2, Guangyuan Liu 2, Guiqin Liu 4, Lingjiang Min 5* and Fuwei Zhao 2* 1 College of Animal Science and Technology, Shandong Agricultural University, Taian, China, 2 National Engineering Research Center for Gelatin-Based Traditional Chinese Medicine, Dong-E E-Jiao Co., Ltd, Liaocheng, China, 3 Institute of Animal Husbandry and Veterinary Medicine, Beijing Academy of Agricultural and Forestry Sciences, Beijing, China, 4 Shandong Donkey Industry, Technology Collaborative Innovation Center, Liaocheng University, Liaocheng, China, 5 College of Animal Science and Technology, Qingdao Agricultural University, Qingdao, China Previous studies have found donkey milk (DM) has the similar compositions with human Edited by: milk (HM) and could be used as a potential hypoallergenic replacement diet for babies Aida Serra, IMDEA Food Institute, Spain suffering from cow’s milk allergy. Milk fat globule membrane (MFGM) proteins are involved Reviewed by: in many biological functions, behaving as important indicators of the nutritional quality of Tânia Rodrigues Dias, milk. In this study, we used label-free proteomics to quantify the differentially expressed University of Aveiro, Portugal MFGM proteins (DEP) between DM (in 4–5 months of lactation) and HM (in 6–8 months Joana Costa, University of Porto, Portugal of lactation). In total, 293 DEP were found in these two groups. Gene Ontology (GO) Céline Henry, enrichment analysis revealed that the majority of DEP participated in regulation of immune INRAE Centre Jouy-en-Josas, France system process, membrane invagination and lymphocyte activation. -
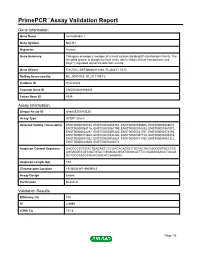
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name nucleobindin 1 Gene Symbol NUCB1 Organism Human Gene Summary This gene encodes a member of a small calcium-binding EF-hand protein family. The encoded protein is thought to have a key role in Golgi calcium homeostasis and Ca(2+)-regulated signal transduction events. Gene Aliases CALNUC, DKFZp686A15286, FLJ40471, NUC RefSeq Accession No. NC_000019.9, NT_011109.16 UniGene ID Hs.631602 Ensembl Gene ID ENSG00000104805 Entrez Gene ID 4924 Assay Information Unique Assay ID qHsaCED0043030 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000304734, ENST00000493763, ENST00000539605, ENST00000538010, ENST00000284116, ENST00000267199, ENST00000324333, ENST00000381971, ENST00000402297, ENST00000397226, ENST00000341761, ENST00000374193, ENST00000313663, ENST00000334126, ENST00000397128, ENST00000405315, ENST00000407032, ENST00000452087, ENST00000411700, ENST00000451312, ENST00000424608, ENST00000263273 Amplicon Context Sequence GACCCCTGCGACTGAGAGTCCCGACACAGGCCTGTACTACCACCGGTACCTCC AGGAGGTCATCGATGTACTGGAGACGGATGGGCATTTCCGAGAGAAGCTGCAG GCTGCCAATGCGGAGGACATCAAGAGC Amplicon Length (bp) 103 Chromosome Location 19:49404167-49409012 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 105 R2 0.9995 cDNA Cq 19.18 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 85 gDNA Cq 24.01 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report NUCB1, Human Amplification Plot -
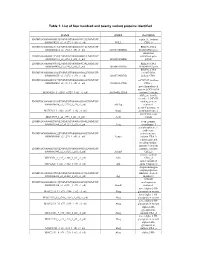
List of Four Hundred and Twenty Variant Proteins Identified
Table 1: List of four hundred and twenty variant proteins identified protein symbol description ENSMUSG00000058013|ENSMUST00000074733|ENSMUSP septin 11, isoform 00000074293_s2_e1277_1_rf0_c1_n0| NULL CRA_b ENSMUSG00000025176|ENSMUST00000081714|ENSMUSP RIKEN cDNA 00000080414_s2_e965_1_rf0_c1_n0| 0610010D20Rik 0610010D20 gene interferon ENSMUSG00000021208|ENSMUST00000044687|ENSMUSP stimulated gene 00000041712_s2_e851_1_rf0_c1_n0| 1810023F06Rik 12(b2) ENSMUSG00000036938|ENSMUST00000064324|ENSMUSP RIKEN cDNA 00000064498_s2_e740_1_rf0_c1_n0| 1810049H19Rik 1810049H19 gene RIKEN cDNA ENSMUSG00000061306|ENSMUST00000045402|ENSMUSP 1810073N04, 00000048675_s2_e3272_1_rf0_c1_n0| 1810073N04Rik isoform CRA_ ENSMUSG00000039879|ENSMUST00000020002|ENSMUSP mCG7517, isoform 00000020002_s2_e314_1_rf0_c1_n0| 3110003A17Rik CRA_c novel hypothetical protein LOC106894 M18C4209_1_s2543_e2729_1_rf1_c1_n0| A630042L21Rik isoform 2 variant ABI gene family, member 3 (NESH) ENSMUSG00000035258|ENSMUST00000048280|ENSMUSP binding protein 00000040830_s2_e2729_1_rf0_c1_n0| Abi3bp isoform 1 acetyl-Coenzyme A M17C923_1_s128_e1337_1_rf2_c1_n0| Acat2 acetyltransferase 2 novel beta actin MXC1985_1_s2_e593_1_rf1_c1_n0| Actb variant ENSMUSG00000029580|ENSMUST00000031564|ENSMUSP actin, gamma, 00000031564_s2_e1127_1_rf0_c1_n0| Actg cytoplasmic 1 acylphosphatase 1, erythrocyte ENSMUSG00000008822|ENSMUST00000008966|ENSMUSP (common) type, 00000008966_s2_e299_1_rf0_c1_n0| Acyp1 isoform CRA_b a disintegrin and metallopeptidase domain 9 (meltrin ENSMUSG00000031555|ENSMUST00000084032|ENSMUSP -

EMP3, a Myelin-Related Gene Located in the Critical 19Q13.3
Priority Reports EMP3, a Myelin-Related Gene Located in the Critical 19q13.3 Region, Is Epigenetically Silenced and Exhibits Features of a Candidate Tumor Suppressor in Glioma and Neuroblastoma Miguel Alaminos,1,2 Vero´nica Da´valos,1 Santiago Ropero,1 Fernando Setie´n,1 Maria F. Paz,1 Michel Herranz,1 Mario F. Fraga,1 Jaume Mora,3 Nai-Kong V. Cheung,4 William L. Gerald,5 and Manel Esteller1 1Cancer Epigenetics Laboratory, Molecular Pathology Programme, Spanish National Cancer Centre, Madrid, Spain; 2Department of Histology, Unidad Mixta de Investigaciones Me´dicas, University Hospital San Cecilio, Granada, Spain; 3Department of Oncology, Hospital Sant Joan de Deu, Barcelona, Spain; and Departments of 4Pediatrics and 5Pathology, Memorial Sloan-Kettering Cancer Center, New York, New York Abstract particularly in adults, usually with a poor prognosis (3). It is The presence of common genomic deletions in the 19q13 thought that both neuroblastomas and gliomas might arise from chromosomal region in neuroblastomas and gliomas strongly neuroectodermal precursor cells (4). suggests the presence of a putative tumor suppressor gene for With an overall survival rate of <50%, the prognosis of these neoplasms in this region that, despite much effort, has neuroblastoma patients is highly variable and is associated with not yet been identified. In an attempt to address this issue, we many biological and clinical features, including the stage of the compared the expression profile of 89 neuroblastoma tumors disease (5), patient age, tumor ploidy (6), and a series of genetic and with that of benign ganglioneuromas by microarray analysis. epigenetic molecular features of the tumor, such as MYCN Probe sets (637 of 62,839) were significantly down-regulated in amplification (5), 1p deletions, 17q gains, and HOXA9 and RARB2 neuroblastoma tumors, including, most importantly, a gene CpG island hypermethylation (7).