PRAC Minutes July 2013
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

18 December 2020 – to Date)
(18 December 2020 – to date) MEDICINES AND RELATED SUBSTANCES ACT 101 OF 1965 (Gazette No. 1171, Notice No. 1002 dated 7 July 1965. Commencement date: 1 April 1966 [Proc. No. 94, Gazette No. 1413] SCHEDULES Government Notice 935 in Government Gazette 31387 dated 5 September 2008. Commencement date: 5 September 2008. As amended by: Government Notice R1230 in Government Gazette 32838 dated 31 December 2009. Commencement date: 31 December 2009. Government Notice R227 in Government Gazette 35149 dated 15 March 2012. Commencement date: 15 March 2012. Government Notice R674 in Government Gazette 36827 dated 13 September 2013. Commencement date: 13 September 2013. Government Notice R690 in Government Gazette 36850 dated 20 September 2013. Commencement date: 20 September 2013. Government Notice R104 in Government Gazette 37318 dated 11 February 2014. Commencement date: 11 February 2014. Government Notice R352 in Government Gazette 37622 dated 8 May 2014. Commencement date: 8 May 2014. Government Notice R234 in Government Gazette 38586 dated 20 March 2015. Commencement date: 20 March 2015. Government Notice 254 in Government Gazette 39815 dated 15 March 2016. Commencement date: 15 March 2016. Government Notice 620 in Government Gazette 40041 dated 3 June 2016. Commencement date: 3 June 2016. Prepared by: Page 2 of 199 Government Notice 748 in Government Gazette 41009 dated 28 July 2017. Commencement date: 28 July 2017. Government Notice 1261 in Government Gazette 41256 dated 17 November 2017. Commencement date: 17 November 2017. Government Notice R1098 in Government Gazette 41971 dated 12 October 2018. Commencement date: 12 October 2018. Government Notice R1262 in Government Gazette 42052 dated 23 November 2018. -

Predictive QSAR Tools to Aid in Early Process Development of Monoclonal Antibodies
Predictive QSAR tools to aid in early process development of monoclonal antibodies John Micael Andreas Karlberg Published work submitted to Newcastle University for the degree of Doctor of Philosophy in the School of Engineering November 2019 Abstract Monoclonal antibodies (mAbs) have become one of the fastest growing markets for diagnostic and therapeutic treatments over the last 30 years with a global sales revenue around $89 billion reported in 2017. A popular framework widely used in pharmaceutical industries for designing manufacturing processes for mAbs is Quality by Design (QbD) due to providing a structured and systematic approach in investigation and screening process parameters that might influence the product quality. However, due to the large number of product quality attributes (CQAs) and process parameters that exist in an mAb process platform, extensive investigation is needed to characterise their impact on the product quality which makes the process development costly and time consuming. There is thus an urgent need for methods and tools that can be used for early risk-based selection of critical product properties and process factors to reduce the number of potential factors that have to be investigated, thereby aiding in speeding up the process development and reduce costs. In this study, a framework for predictive model development based on Quantitative Structure- Activity Relationship (QSAR) modelling was developed to link structural features and properties of mAbs to Hydrophobic Interaction Chromatography (HIC) retention times and expressed mAb yield from HEK cells. Model development was based on a structured approach for incremental model refinement and evaluation that aided in increasing model performance until becoming acceptable in accordance to the OECD guidelines for QSAR models. -

Modifications to the Harmonized Tariff Schedule of the United States To
U.S. International Trade Commission COMMISSIONERS Shara L. Aranoff, Chairman Daniel R. Pearson, Vice Chairman Deanna Tanner Okun Charlotte R. Lane Irving A. Williamson Dean A. Pinkert Address all communications to Secretary to the Commission United States International Trade Commission Washington, DC 20436 U.S. International Trade Commission Washington, DC 20436 www.usitc.gov Modifications to the Harmonized Tariff Schedule of the United States to Implement the Dominican Republic- Central America-United States Free Trade Agreement With Respect to Costa Rica Publication 4038 December 2008 (This page is intentionally blank) Pursuant to the letter of request from the United States Trade Representative of December 18, 2008, set forth in the Appendix hereto, and pursuant to section 1207(a) of the Omnibus Trade and Competitiveness Act, the Commission is publishing the following modifications to the Harmonized Tariff Schedule of the United States (HTS) to implement the Dominican Republic- Central America-United States Free Trade Agreement, as approved in the Dominican Republic-Central America- United States Free Trade Agreement Implementation Act, with respect to Costa Rica. (This page is intentionally blank) Annex I Effective with respect to goods that are entered, or withdrawn from warehouse for consumption, on or after January 1, 2009, the Harmonized Tariff Schedule of the United States (HTS) is modified as provided herein, with bracketed matter included to assist in the understanding of proclaimed modifications. The following supersedes matter now in the HTS. (1). General note 4 is modified as follows: (a). by deleting from subdivision (a) the following country from the enumeration of independent beneficiary developing countries: Costa Rica (b). -

The Two Tontti Tudiul Lui Hi Ha Unit
THETWO TONTTI USTUDIUL 20170267753A1 LUI HI HA UNIT ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2017 /0267753 A1 Ehrenpreis (43 ) Pub . Date : Sep . 21 , 2017 ( 54 ) COMBINATION THERAPY FOR (52 ) U .S . CI. CO - ADMINISTRATION OF MONOCLONAL CPC .. .. CO7K 16 / 241 ( 2013 .01 ) ; A61K 39 / 3955 ANTIBODIES ( 2013 .01 ) ; A61K 31 /4706 ( 2013 .01 ) ; A61K 31 / 165 ( 2013 .01 ) ; CO7K 2317 /21 (2013 . 01 ) ; (71 ) Applicant: Eli D Ehrenpreis , Skokie , IL (US ) CO7K 2317/ 24 ( 2013. 01 ) ; A61K 2039/ 505 ( 2013 .01 ) (72 ) Inventor : Eli D Ehrenpreis, Skokie , IL (US ) (57 ) ABSTRACT Disclosed are methods for enhancing the efficacy of mono (21 ) Appl. No. : 15 /605 ,212 clonal antibody therapy , which entails co - administering a therapeutic monoclonal antibody , or a functional fragment (22 ) Filed : May 25 , 2017 thereof, and an effective amount of colchicine or hydroxy chloroquine , or a combination thereof, to a patient in need Related U . S . Application Data thereof . Also disclosed are methods of prolonging or increasing the time a monoclonal antibody remains in the (63 ) Continuation - in - part of application No . 14 / 947 , 193 , circulation of a patient, which entails co - administering a filed on Nov. 20 , 2015 . therapeutic monoclonal antibody , or a functional fragment ( 60 ) Provisional application No . 62/ 082, 682 , filed on Nov . of the monoclonal antibody , and an effective amount of 21 , 2014 . colchicine or hydroxychloroquine , or a combination thereof, to a patient in need thereof, wherein the time themonoclonal antibody remains in the circulation ( e . g . , blood serum ) of the Publication Classification patient is increased relative to the same regimen of admin (51 ) Int . -

(12) Patent Application Publication (10) Pub. No.: US 2017/0172932 A1 Peyman (43) Pub
US 20170172932A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0172932 A1 Peyman (43) Pub. Date: Jun. 22, 2017 (54) EARLY CANCER DETECTION AND A 6LX 39/395 (2006.01) ENHANCED IMMUNOTHERAPY A61R 4I/00 (2006.01) (52) U.S. Cl. (71) Applicant: Gholam A. Peyman, Sun City, AZ CPC .......... A61K 9/50 (2013.01); A61K 39/39558 (US) (2013.01); A61K 4I/0052 (2013.01); A61 K 48/00 (2013.01); A61K 35/17 (2013.01); A61 K (72) Inventor: sham A. Peyman, Sun City, AZ 35/15 (2013.01); A61K 2035/124 (2013.01) (21) Appl. No.: 15/143,981 (57) ABSTRACT (22) Filed: May 2, 2016 A method of therapy for a tumor or other pathology by administering a combination of thermotherapy and immu Related U.S. Application Data notherapy optionally combined with gene delivery. The combination therapy beneficially treats the tumor and pre (63) Continuation-in-part of application No. 14/976,321, vents tumor recurrence, either locally or at a different site, by filed on Dec. 21, 2015. boosting the patient’s immune response both at the time or original therapy and/or for later therapy. With respect to Publication Classification gene delivery, the inventive method may be used in cancer (51) Int. Cl. therapy, but is not limited to such use; it will be appreciated A 6LX 9/50 (2006.01) that the inventive method may be used for gene delivery in A6 IK 35/5 (2006.01) general. The controlled and precise application of thermal A6 IK 4.8/00 (2006.01) energy enhances gene transfer to any cell, whether the cell A 6LX 35/7 (2006.01) is a neoplastic cell, a pre-neoplastic cell, or a normal cell. -

Hexoprenaline: Β-Adrenoreceptor Selectivity in Isolated Tissues from the Guinea-Pig
Clinical and Experimental Pharmacology and Physiology (1975) 2, 541-547. Hexoprenaline: p-adrenoreceptor selectivity in isolated tissues from the guinea-pig Stella R. O’Donnell and Janet C. Wanstall Department of Physiology, University of Queenstand, Brisbane, Australia (Received 12 March 1975; revision received 22 April 1975) SUMMARY 1. A catecholamine P-adrenoreceptor agonist, hexoprenaline, was examined in vitro on five guinea-pig tissues and its potency relative to isoprenaline (as 100) obtained. 2. Hexoprenaline clearly delineated between those tissues classified as containing P,-adrenoreceptors (trachea, hind limb blood vessels and uterus; relative potencies 219, 110 and 76 respectively) and those classified as containing P,-adrenoreceptors (atria and ileum; relative potencies 3.3 and 1.0 respectively). 3. Hexoprenaline differed from some previously studied noncatecholamine P-adrenoreceptor agonists in being only two-fold less potent, relative to isoprena- line, as a vasodilator in perfused hind limb than as a tracheal relaxant. Key words : j3-adrenoreceptors, blood vessels, bronchodilators, guinea-pig, hexo- prenaline, selectivity, trachea. INTRODUCTION In a previous study using in vitro preparations from the guinea-pig, O’Donnell & Wanstall (1974a) examined potential sympathomimetic bronchodilator compounds for their potency as tracheal relaxants (p,-adrenoreceptors), atrial stimulants (P,-adrenoreceptors) and vasodilators in the perfused hind limb (p2-adrenoreceptors). Some resorcinolamines showed selectivity for trachea compared with not only atria but also blood vessels. There is some evidence in dogs in vivo that other noncatecholamines, namely carbuterol (Wardell et al., 1974), and salbutamol and terbutaline (Wasserman & Levy, 1974), display similar select- ivity. If selectivity between respiratory and vascular smooth muscle represents selectivity at the p-adrenoreceptor level, it poses the question whether the P1/p2subclassification (Lands et al., 1967a; Lands, Luduena & Buzzo, 1967b) is too rigid. -

WO 2016/176089 Al 3 November 2016 (03.11.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/176089 Al 3 November 2016 (03.11.2016) P O P C T (51) International Patent Classification: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, A01N 43/00 (2006.01) A61K 31/33 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/US2016/028383 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 20 April 2016 (20.04.2016) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/154,426 29 April 2015 (29.04.2015) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: KARDIATONOS, INC. [US/US]; 4909 DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, Lapeer Road, Metamora, Michigan 48455 (US). -
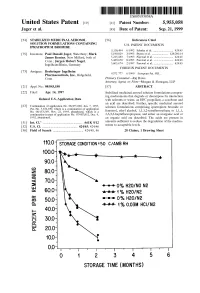
100 Storage Condition=50 C/Ambrh
USOO595.5058A United States Patent (19) 11 Patent Number: S.9SS,0589 9 Jager et al. (45) Date of Patent: Sep. 21,9 1999 54). STABILIZED MEDICINAL AEROSOL 56) References Cited SOLUTION FORMULATIONS CONTAINING U.S. PATENT DOCUMENTS IPRATROPIUM BROMIDE a 5,118,494 6/1992 Schultz et al. ............................ 424/45 75 Inventors: Paul Donald Jager, Waterbury; Mark 5,190,029 3/1993 Byron et al. ... ... 128/200.14 James Kontny, New Milford, both of 5,225,183 7/1993 Purewal et al. ........................... 424/45 Conn.; Jurgen Hubert Nagel 5.439,670 8/1995 Purewal et al. ... 424/45 Ingelheim/Rhein, Germany s 5,605,674 2/1997 Purewal et al. ........................... 424/45 FOREIGN PATENT DOCUMENTS 73 Assignee: Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, 0372 777 6/1990 European Pat. Off.. Conn. Primary Examiner Raj Bawa Attorney, Agent, or Firm Morgan & Finnegan, LLP 21 Appl.pp No.: 08/843,180 57 ABSTRACT 22 Filed: Apr. 14, 1997 Stabilized medicinal aeroSol Solution formulations compris O O ing medicaments that degrade or decompose by interaction Related U.S. Application Data with solvents or water, an HFC propellant, a cosolvent and an acid are described. Further, Specific medicinal aeroSol 63 Staggypt.NE "A iGs Solution formulations comprising ipratropium bromide or No. 08/153.549, Nov. 22, 1993, abandoned E. is a fenoterol, ethyl alcohol, 1,1,1,2-tetrafluoroethane or 1,1,1, continuation-in-part of application No. 07/987,852, Dec. 9, 2,3,3,3-heptafluoropropane, and either an inorganic acid or 1992, abandoned. an organic acid are described. The acids are present in 51 Int. -

L:\0901 with Peru\0901PHARMAPPX.Wpd
Harmonized Tariff Schedule of the United States (2009) (Rev. 1) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2009) (Rev. 1) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. ABACAVIR 136470-78-5 ACEXAMIC ACID 57-08-9 ABAFUNGIN 129639-79-8 ACICLOVIR 59277-89-3 ABAMECTIN 65195-55-3 ACIFRAN 72420-38-3 ABANOQUIL 90402-40-7 ACIPIMOX 51037-30-0 ABAPERIDONE 183849-43-6 ACITAZANOLAST 114607-46-4 ABARELIX 183552-38-7 ACITEMATE 101197-99-3 ABATACEPT 332348-12-6 ACITRETIN 55079-83-9 ABCIXIMAB 143653-53-6 ACIVICIN 42228-92-2 ABECARNIL 111841-85-1 ACLANTATE 39633-62-0 ABETIMUS 167362-48-3 ACLARUBICIN 57576-44-0 ABIRATERONE 154229-19-3 ACLATONIUM NAPADISILATE 55077-30-0 ABITESARTAN 137882-98-5 ACODAZOLE 79152-85-5 ABLUKAST 96566-25-5 ACOLBIFENE 182167-02-8 ABRINEURIN 178535-93-8 ACONIAZIDE 13410-86-1 ABUNIDAZOLE 91017-58-2 ACOTIAMIDE 185106-16-5 ACADESINE 2627-69-2 ACOXATRINE 748-44-7 ACAMPROSATE 77337-76-9 ACREOZAST 123548-56-1 ACAPRAZINE 55485-20-6 -

Addition of Intravenous Beta2-Agonists to Inhaled Beta2- Agonists for Acute Asthma (Review)
Addition of intravenous beta2-agonists to inhaled beta2- agonists for acute asthma (Review) Travers AH, Milan SJ, Jones AP, Camargo Jr CA, Rowe BH This is a reprint of a Cochrane review, prepared and maintained by The Cochrane Collaboration and published in The Cochrane Library 2012, Issue 12 http://www.thecochranelibrary.com Addition of intravenous beta2-agonists to inhaled beta2-agonists for acute asthma (Review) Copyright © 2012 The Cochrane Collaboration. Published by John Wiley & Sons, Ltd. TABLE OF CONTENTS HEADER....................................... 1 ABSTRACT ...................................... 1 PLAINLANGUAGESUMMARY . 2 SUMMARY OF FINDINGS FOR THE MAIN COMPARISON . ..... 3 BACKGROUND .................................... 5 OBJECTIVES ..................................... 5 METHODS ...................................... 5 RESULTS....................................... 7 Figure1. ..................................... 8 Figure2. ..................................... 9 Figure3. ..................................... 10 DISCUSSION ..................................... 12 AUTHORS’CONCLUSIONS . 12 ACKNOWLEDGEMENTS . 13 REFERENCES ..................................... 13 CHARACTERISTICSOFSTUDIES . 19 DATAANDANALYSES. 31 Analysis 1.1. Comparison 1 IV + inhaled beta agonist vs. inhaled beta-agonist, Outcome 1 Admissions. 31 Analysis 1.2. Comparison 1 IV + inhaled beta agonist vs. inhaled beta-agonist, Outcome 2 Length of stay. 32 Analysis 1.3. Comparison 1 IV + inhaled beta agonist vs. inhaled beta-agonist, Outcome 3 Pulse rate at 2 -

210428Orig1s000
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 210428Orig1s000 OTHER REVIEW(S) DIVISION OF CARDIOVASCULAR AND RENAL PRODUCTS Regulatory Project Manager Overview I. GENERAL INFORMATION NDA: 210428 Drug: Metoprolol Succinate Extended-Release Capsules, 25 mg, 50 mg, 100 mg and 200 mg Class: Beta- Blocker Applicant: Sun Pharmaceutical Industries Limited Proposed Indications: Hypertension, Angina Pectoris & Heart Failure Date of submission: March 30, 2017 PDUFA date: January 30, 2018 II. REVIEW TEAM Office of New Drugs, Office of Drug Evaluation I: Division of Cardiovascular & Renal Product Norman Stockbridge, MD, PhD, Director Mary Ross Southworth, PharmD, Deputy Director for Safety Michael Monteleone, MS, RAC, Assistant Director for Labeling Martina Sahre, PhD, Cross Discipline Team Leader Fortunato Senatore, MD, Clinical Reviewer Albert DeFelice, PhD, Non-Clinical Supervisor Muriel Saulnier, PhD, Non-Clinical Reviewer Edward Fromm, RPh, RAC, Chief, Project Management Staff Maryam Changi, PharmD, Regulatory Project Manager Office of Pharmaceutical Quality: Wendy Wilson-Lee, PhD, Application Technical Lead Grafton Adams, Regulatory Business Process Manager Milton Sloan, PhD, Drug Product Reviewer Rohit Tiwari, PhD, Drug Substance Reviewer Ben Stevens, PhD, Drug Substance Team Leader Kaushal Dave, PhD, Biopharmaceutical Reviewer Wu, Ta-Chin, PhD, Biopharmaceutical Team Leader Viviana Matta, PhD, Facility Reviewer Ruth Moore, PhD, Facility Team Leader Chunsheng Cai, PhD, Process Reviewer NDA 210428 RPM review Page 1 Reference ID: 42132034213643 Labeling/PMC-PMR to Sponsor: November 30, 2017 PDUFA Date: January 30, 2018 (Standard, 10-Month) PDUFA Goal Date: January 30, 2018 Approval letter: January 26, 2018 7. Reviews a) Divisional Memorandum: (January 26, 2018) Dr. Stockbridge indicated his concurrence on Dr. Sahre’s CDTL memo. -

An Update on Chronic Obstructive Pulmonary Disease
South African Family Practice 2016; 58(3):6-12 S Afr Fam Pract Open Access article distributed under the terms of the ISSN 2078-6190 EISSN 2078-6204 Creative Commons License [CC BY-NC-ND 4.0] © 2016 The Author(s) http://creativecommons.org/licenses/by-nc-nd/4.0 REVIEW An update on chronic obstructive pulmonary disease Natalie Schellack,1 Gustav Schellack,2 Richard Omoding1 1Department of Pharmacy, Faculty of Health Sciences, Sefako Makgatho Health Sciences University 2Pharmaceutical Industry Corresponding author, email: [email protected] Abstract Chronic obstructive pulmonary disease (COPD) is a leading cause of death worldwide. It is a chronic condition which affects the respiratory system and worsens over time. Cigarette smoking and advancing age are the two major risks associated with this disease. It is concerning that the global incidence of this chronic illness is on the rise. Current projections indicate that it will become the third leading cause of death by the year 2020. Inflammatory changes underlie the pathophysiology of COPD. Irreversible damage and progressive narrowing of the air passages follow. COPD is characterised by the progressive loss of lung function. In addition, the Global Initiative for Chronic Obstructive Lung Disease released the latest update on its global strategy for the diagnosis, management, and prevention of COPD in 2015. This article provides an overview of the causative risk factors, underlying disease process, pathophysiological changes, and the classification and management of COPD, including the latest perspectives on this highly prevalent condition. Keywords: chronic bronchitis, chronic obstructive pulmonary disease, COPD, emphysema, LABA, SABA, SAMA, LAMA, methylxanthines Introduction Aetiology and pathogenesis Chronic obstructive pulmonary disease (COPD) is a leading cause of death worldwide.