Ep 1931310 B1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Appendix 1 Table of Contents
@ECHA EUROPEAN CHEMICALS AGENCY Appendix 1 Table of Contents: Supplementary Table C:Substances on Annex II of the Cosmetic Products Regulation ... 1 Supplementary Table D: Substances proposed to be restricted due their use restriction in CPR Annex IV column 9..,,.. 119 Supplementary Table E: Substances allowed in tattoo inks subject to the conditions specified in CPR Annex IV columns h and i ......r29 Annankatu 18. P.O. Box 400, FI-00121 Helsinki, Finland I Tel. +358 9 686180 | Fax +358 9 68618210 | echa.europa.eu ANNEX XV RESTRICTION REPORT - SUBSTANCES IN TATTOO INKS AND PERMANENT MAKE UP Su lemen Table C:Substances on Annex II of the Cosmetic Products Re ulationl Substance EC# cAs # Substance EC# cAs # R T T T A- s c R I T Name Name e b b b II s M 7 c I s I I I #4 5 6 D 9 2 1 2 3 a 3 3 3 N-(s- Chlorobenzoxa zol-2- 35783- vl)acetamide 57-4 1 (2- Acetoxyethyl)t rimethylammo (2- nium acetoxyethyl hydroxide )trimethyla 200- (Acetylcholine) 200- mmonium 124-9 5 1-84-3 and its salts t2a-9 51-84-3 2 Deanol Deanol 222- 3342- aceglumate 222- 3342- aceqlumate 085-5 61-8 (INN) 085-5 61-8 3 Spironolacto 200- Spironolactone 200- ne 133-6 52-O1-7 rINN) 133-6 52-0L-7 4 14-(4- Hydroxy-3- iodophenoxy)- 3,5- diiodophenylla cetic acid (Tiratricol 200- (INN)) and its 200- Tiratricol 086- 1 5r-24-7 salts 086- 1 5l-24-7 5 Methotrexat 200- Methotrexate 200- e 413-8 59-05-2 (INN) 413-8 59-05-2 6 Aminocaproic Aminocaproi 200- acid (INN) and 200- c acid 469-3 60-32-2 its salts 469-3 60-32-2 7 Cinchophen (rNN), its salts, derivatives and salts of 205- 132-60- these 205- 132-60- Cinchophen 067-r 5 derivatives 067-l 5 Thyropropic acid (INN) and its salts 5L-26-3 9 Trichloroacet 200- Trichloroacetic 200- ic acid 927-2 75-03-9 acid 927-2 76-03-9 l0 Aconitum napellus L. -

The National Drugs List
^ ^ ^ ^ ^[ ^ The National Drugs List Of Syrian Arab Republic Sexth Edition 2006 ! " # "$ % &'() " # * +$, -. / & 0 /+12 3 4" 5 "$ . "$ 67"5,) 0 " /! !2 4? @ % 88 9 3: " # "$ ;+<=2 – G# H H2 I) – 6( – 65 : A B C "5 : , D )* . J!* HK"3 H"$ T ) 4 B K<) +$ LMA N O 3 4P<B &Q / RS ) H< C4VH /430 / 1988 V W* < C A GQ ") 4V / 1000 / C4VH /820 / 2001 V XX K<# C ,V /500 / 1992 V "!X V /946 / 2004 V Z < C V /914 / 2003 V ) < ] +$, [2 / ,) @# @ S%Q2 J"= [ &<\ @ +$ LMA 1 O \ . S X '( ^ & M_ `AB @ &' 3 4" + @ V= 4 )\ " : N " # "$ 6 ) G" 3Q + a C G /<"B d3: C K7 e , fM 4 Q b"$ " < $\ c"7: 5) G . HHH3Q J # Hg ' V"h 6< G* H5 !" # $%" & $' ,* ( )* + 2 ا اوا ادو +% 5 j 2 i1 6 B J' 6<X " 6"[ i2 "$ "< * i3 10 6 i4 11 6! ^ i5 13 6<X "!# * i6 15 7 G!, 6 - k 24"$d dl ?K V *4V h 63[46 ' i8 19 Adl 20 "( 2 i9 20 G Q) 6 i10 20 a 6 m[, 6 i11 21 ?K V $n i12 21 "% * i13 23 b+ 6 i14 23 oe C * i15 24 !, 2 6\ i16 25 C V pq * i17 26 ( S 6) 1, ++ &"r i19 3 +% 27 G 6 ""% i19 28 ^ Ks 2 i20 31 % Ks 2 i21 32 s * i22 35 " " * i23 37 "$ * i24 38 6" i25 39 V t h Gu* v!* 2 i26 39 ( 2 i27 40 B w< Ks 2 i28 40 d C &"r i29 42 "' 6 i30 42 " * i31 42 ":< * i32 5 ./ 0" -33 4 : ANAESTHETICS $ 1 2 -1 :GENERAL ANAESTHETICS AND OXYGEN 4 $1 2 2- ATRACURIUM BESYLATE DROPERIDOL ETHER FENTANYL HALOTHANE ISOFLURANE KETAMINE HCL NITROUS OXIDE OXYGEN PROPOFOL REMIFENTANIL SEVOFLURANE SUFENTANIL THIOPENTAL :LOCAL ANAESTHETICS !67$1 2 -5 AMYLEINE HCL=AMYLOCAINE ARTICAINE BENZOCAINE BUPIVACAINE CINCHOCAINE LIDOCAINE MEPIVACAINE OXETHAZAINE PRAMOXINE PRILOCAINE PREOPERATIVE MEDICATION & SEDATION FOR 9*: ;< " 2 -8 : : SHORT -TERM PROCEDURES ATROPINE DIAZEPAM INJ. -

Classification of Medicinal Drugs and Driving: Co-Ordination and Synthesis Report
Project No. TREN-05-FP6TR-S07.61320-518404-DRUID DRUID Driving under the Influence of Drugs, Alcohol and Medicines Integrated Project 1.6. Sustainable Development, Global Change and Ecosystem 1.6.2: Sustainable Surface Transport 6th Framework Programme Deliverable 4.4.1 Classification of medicinal drugs and driving: Co-ordination and synthesis report. Due date of deliverable: 21.07.2011 Actual submission date: 21.07.2011 Revision date: 21.07.2011 Start date of project: 15.10.2006 Duration: 48 months Organisation name of lead contractor for this deliverable: UVA Revision 0.0 Project co-funded by the European Commission within the Sixth Framework Programme (2002-2006) Dissemination Level PU Public PP Restricted to other programme participants (including the Commission x Services) RE Restricted to a group specified by the consortium (including the Commission Services) CO Confidential, only for members of the consortium (including the Commission Services) DRUID 6th Framework Programme Deliverable D.4.4.1 Classification of medicinal drugs and driving: Co-ordination and synthesis report. Page 1 of 243 Classification of medicinal drugs and driving: Co-ordination and synthesis report. Authors Trinidad Gómez-Talegón, Inmaculada Fierro, M. Carmen Del Río, F. Javier Álvarez (UVa, University of Valladolid, Spain) Partners - Silvia Ravera, Susana Monteiro, Han de Gier (RUGPha, University of Groningen, the Netherlands) - Gertrude Van der Linden, Sara-Ann Legrand, Kristof Pil, Alain Verstraete (UGent, Ghent University, Belgium) - Michel Mallaret, Charles Mercier-Guyon, Isabelle Mercier-Guyon (UGren, University of Grenoble, Centre Regional de Pharmacovigilance, France) - Katerina Touliou (CERT-HIT, Centre for Research and Technology Hellas, Greece) - Michael Hei βing (BASt, Bundesanstalt für Straßenwesen, Germany). -

Hyperinflation Management Medications Requiring Prior Authorization for Medical Necessity
July 2021 Effective 07/01/2021 Hyperinflation Management Medications Requiring Prior Authorization for Medical Necessity Below is a list of medicines by drug class that will not be covered without a prior authorization for medical necessity. If you continue using one of these drugs without prior approval for medical necessity, you may be required to pay the full cost. If you are currently using one of the drugs requiring prior authorization for medical necessity, ask your d octor to choose one of the generic or brand formulary options listed below. Category * Drugs Requiring Prior Formulary Options Drug Class Authorization for Medical Necessity 1 Allergies Dexchlorpheniramine levocetirizine Antihistamines Diphen Elixir (NDC^ 69067009204 only) RyClora CARBINOXAMINE TABLET 6 MG Anti-convulsants topiramate ext-rel capsule carbamazepine, carbamazepine ext-rel, (generics for QUDEXY XR only) clobazam, divalproex sodium, divalproex sodium ext-rel, gabapentin, lamotrigine, lamotrigine ext-rel, levetiracetam, levetiracetam ext-rel, oxcarbazepine, phenobarbital, phenytoin, phenytoin sodium extended, primidone, rufinamide, tiagabine, topiramate, valproic acid, zonisamide, FYCOMPA, OXTELLAR XR, TROKENDI XR, VIMPAT, XCOPRI ZONEGRAN carbamazepine, carbamazepine ext-rel, divalproex sodium, divalproex sodium ext-rel, gabapentin, lamotrigine, lamotrigine ext-rel, levetiracetam, levetiracetam ext-rel, oxcarbazepine, phenobarbital, phenytoin, phenytoin sodium extended, primidone, tiagabine, topiramate, valproic acid, zonisamide, FYCOMPA, OXTELLAR XR, TROKENDI -

Medicines Used Across Wirral Health Economy
Medicines used across Wirral Health Economy Below is a list of medicines that are approved for use on Wirral. Some medicines may only be prescribed in hospital, others may be prescribed by your GP. The list is updated every time a new medicine is approved for use in Wirral. Some medicines have been recommended by the National Institute for Health and Care Excellence (NICE). The middle column of the table below indicates recommendations made by NICE — eg, TA234 means that the medicine was reviewed in NICE technology appraisal number 234. Some of the medicines we use are recommended by specialists working from the Walton Centre NHS Foundation Trust or from the Cheshire and Wirral Partnership NHS Foundation Trust. These medicines are included in our formulary — however; the prescriber retains responsibility for the appropriate use of these medicines. These formularies are obtainable here: http://www.panmerseyapc.nhs.uk/formulary/documents/04- 0800_antiepileptic_drugs.pdf http://www.panmerseyapc.nhs.uk/formulary/documents/04-09- 00_parkinsonism.pdf http://www.cwp.nhs.uk/pages/629-pharmacy-and-medicines-information Subject to NICE Drug Name recommendation Abatacept TA195, TA280 Acamprosate Acarbose Accu-Chek Inform II® AccuSol® 35 Potassium 4mmol/L Acetazolamide Acetylcysteine Acencoumarol Acetic acid 5% solution Acetone Acitretin Aciclovir Wirral Medicines Formulary Author: Medicines Management Team Approved by: Wirral Drug and Therapeutics Panel Last updated: June 2015 Version 10 Aclinidium Actilite® Activated Charcoal Activon® - see -
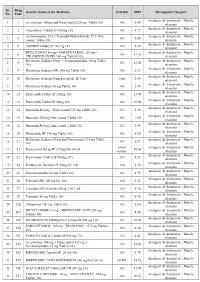
PMBJP Product.Pdf
Sr. Drug Generic Name of the Medicine Unit Size MRP Therapeutic Category No. Code Analgesic & Antipyretic / Muscle 1 1 Aceclofenac 100mg and Paracetamol 325 mg Tablet 10's 10's 8.00 relaxants Analgesic & Antipyretic / Muscle 2 2 Aceclofenac Tablets IP 100mg 10's 10's 4.37 relaxants Acetaminophen 325 + Tramadol Hydrochloride 37.5 film Analgesic & Antipyretic / Muscle 3 4 10's 8.00 coated Tablet 10's relaxants Analgesic & Antipyretic / Muscle 4 5 ASPIRIN Tablets IP 150 mg 14's 14's 2.70 relaxants DICLOFENAC 50 mg+ PARACETAMOL 325 mg+ Analgesic & Antipyretic / Muscle 5 6 10's 11.30 CHLORZOXAZONE 500 mg Tablets 10's relaxants Diclofenac Sodium 50mg + Serratiopeptidase 10mg Tablet Analgesic & Antipyretic / Muscle 6 8 10's 12.00 10's relaxants Analgesic & Antipyretic / Muscle 7 9 Diclofenac Sodium (SR) 100 mg Tablet 10's 10's 6.12 relaxants Analgesic & Antipyretic / Muscle 8 10 Diclofenac Sodium 25mg per ml Inj. IP 3 ml 3 ml 2.00 relaxants Analgesic & Antipyretic / Muscle 9 11 Diclofenac Sodium 50 mg Tablet 10's 10's 2.90 relaxants Analgesic & Antipyretic / Muscle 10 12 Etoricoxilb Tablets IP 120mg 10's 10's 33.00 relaxants Analgesic & Antipyretic / Muscle 11 13 Etoricoxilb Tablets IP 90mg 10's 10's 25.00 relaxants Analgesic & Antipyretic / Muscle 12 14 Ibuprofen 400 mg + Paracetamol 325 mg Tablet 10's 15's 5.50 relaxants Analgesic & Antipyretic / Muscle 13 15 Ibuprofen 200 mg film coated Tablet 10's 10's 1.80 relaxants Analgesic & Antipyretic / Muscle 14 16 Ibuprofen 400 mg film coated Tablet 10's 15's 3.50 relaxants Analgesic & Antipyretic -

Pdf; Chi 2015 DPP Air in Cars.Pdf; Dodson 2014 DPP Dust CA.Pdf; Kasper-Sonnenberg 2014 Phth Metabolites.Pdf; EU Cosmetics Regs 2009.Pdf
Bouge, Cathy (ECY) From: Nancy Uding <[email protected]> Sent: Friday, January 13, 2017 10:24 AM To: Steward, Kara (ECY) Cc: Erika Schreder Subject: Comments re. 2016 CSPA Rule Update - DPP Attachments: DPP 131-18-0 exposure.pdf; Chi 2015 DPP air in cars.pdf; Dodson 2014 DPP dust CA.pdf; Kasper-Sonnenberg 2014 phth metabolites.pdf; EU Cosmetics Regs 2009.pdf Please accept these comments from Toxic-Free Future concerning the exposure potential of DPP for consideration during the 2016 CSPA Rule update. Regards, Nancy Uding -- Nancy Uding Grants & Research Specialist Toxic-Free Future 206-632-1545 ext.123 http://toxicfreefuture.org 1 JES-00888; No of Pages 9 JOURNAL OF ENVIRONMENTAL SCIENCES XX (2016) XXX– XXX Available online at www.sciencedirect.com ScienceDirect www.elsevier.com/locate/jes Determination of 15 phthalate esters in air by gas-phase and particle-phase simultaneous sampling Chenchen Chi1, Meng Xia1, Chen Zhou1, Xueqing Wang1,2, Mili Weng1,3, Xueyou Shen1,4,⁎ 1. College of Environmental & Resource Sciences, Zhejiang University, Hangzhou 310058, China 2. Zhejiang National Radiation Environmental Technology Co., Ltd., Hangzhou 310011, China 3. School of Environmental and Resource Sciences, Zhejiang Agriculture and Forestry University, Hangzhou 310058, China 4. Zhejiang Provincial Key Laboratory of Organic Pollution Process and Control, Hangzhou 310058, China ARTICLE INFO ABSTRACT Article history: Based on previous research, the sampling and analysis methods for phthalate esters (PAEs) Received 24 December 2015 were improved by increasing the sampling flow of indoor air from 1 to 4 L/min, shortening the Revised 14 January 2016 sampling duration from 8 to 2 hr. -

Meta-Xylene: Identification of a New Antigenic Entity in Hypersensitivity
Clinical Communications Meta-xylene: identification of a new Our patient is a 53-year-old male who presented in 2003 with antigenic entity in hypersensitivity contact dermatitis after the use of lidocaine and disinfection with reactions to local anesthetics chlorhexidine. In 2006, an extensive skin testing by patch, prick, Kuntheavy Ing Lorenzini, PhDa, intradermal sampling, and graded challenge was performed as b c followed. The patch tests were performed with the thin layer Fabienne Gay-Crosier Chabry, MD , Claude Piguet, PhD , rapid use epicutaneous test, using the caine mix (benzocaine, a and Jules Desmeules, MD tetracaine, and cinchocaine), the paraben mix (methyl, ethyl, propyl, butyl, and benzylparaben), and the Balsam of Peru. The Clinical Implications caine and paraben mix were positive, and the patch test was possibly positive for Balsam of Peru. The prick test performed The authors report a patient who presented delayed with preservative-free lidocaine 20 mg/mL (Lidocaïne HCl hypersensitivity reactions to several local anesthetics, all “Bichsel” 2%, Grosse Apotheke Dr. G. Bichsel, Switzerland) was containing a meta-xylene entity, but not to articaine, negative. The intradermal test, performed with lidocaine 20 mg/ which is a thiophene derivative. Meta-xylene could be the mL containing methylparaben (Xylocain 2%, AstraZeneca, immunogenic epitope. Switzerland) with 1/10 and 1/100 dilutions, was negative. The subcutaneous challenge test was performed with 10 mg of preservative-free lidocaine 20 mg/mL (Lidocaïne HCl “Bichsel” TO THE EDITOR: 2%, Grosse Apotheke Dr. G. Bichsel) and lidocaine 20 mg/mL containing methylparaben (Lidocaïne Streuli 2%, Streuli, Hypersensitivity reactions to local anesthetics (LAs) are rare and Switzerland), both of which were positive and had the appear- represent less than 1% of all adverse reactions to LAs. -

Col.V.MISSION of the EUROPEAN COMMUNITIES
COl.V.MISSION OF THE EUROPEAN COMMUNITIES SEC(90) 1985 final· Brussels, 29 October 1990 Proposa I for a COUNCIL DIRECTIVE on the approximation of the laws of the Member States relat.ing to cosmetic products -2- EXPLANATORY UEUORANDUM 1. In the context of a people's Europe, the Commission attaches great Importance to simplifying and clarifying Community law so as to make It clearer and more accessible to the ordinary citizen, thus giving hIm new opportunItIes and the chance to make use of the spec if i c rights It gives him. This aim cannot be achieved so long as numerous provisions that have been amended several times, often QUite substantially, remain scattered, so that they must be sought partly In the original Instrument and partly In later amending ones. Considerable research work, comparing many different Instruments, Is thus needed to Identify the current rules. For this reason a consot tdatton of rules that have freQuently been amended Is essential If Community law Is to be clear and transparent. 2. In Its resolution of 26 November 1974 concerning consolidation of Its acts (1), the Council recommended that those of Its acts which have been amended several times be assembled Into a single text. It stressed that, In the Interests of legal certainty, a genuine legislative consolidation, Involving the repeal of earlier acts, should wherever possible be effected (as Is being done In this case). it conseQuently Invited the Commission to let it have proposals for consol !dation and undertook ·to examine them "as Quickly as possible, whltout bringing Into QUestion, during that consol ldatlon, the substantive solutions contained In the consol !dated texts". -

1: Gastro-Intestinal System
1 1: GASTRO-INTESTINAL SYSTEM Antacids .......................................................... 1 Stimulant laxatives ...................................46 Compound alginate products .................. 3 Docuate sodium .......................................49 Simeticone ................................................... 4 Lactulose ....................................................50 Antimuscarinics .......................................... 5 Macrogols (polyethylene glycols) ..........51 Glycopyrronium .......................................13 Magnesium salts ........................................53 Hyoscine butylbromide ...........................16 Rectal products for constipation ..........55 Hyoscine hydrobromide .........................19 Products for haemorrhoids .................56 Propantheline ............................................21 Pancreatin ...................................................58 Orphenadrine ...........................................23 Prokinetics ..................................................24 Quick Clinical Guides: H2-receptor antagonists .......................27 Death rattle (noisy rattling breathing) 12 Proton pump inhibitors ........................30 Opioid-induced constipation .................42 Loperamide ................................................35 Bowel management in paraplegia Laxatives ......................................................38 and tetraplegia .....................................44 Ispaghula (Psyllium husk) ........................45 ANTACIDS Indications: -

Local Anesthetics
Local Anesthetics Introduction and History Cocaine is a naturally occurring compound indigenous to the Andes Mountains, West Indies, and Java. It was the first anesthetic to be discovered and is the only naturally occurring local anesthetic; all others are synthetically derived. Cocaine was introduced into Europe in the 1800s following its isolation from coca beans. Sigmund Freud, the noted Austrian psychoanalyst, used cocaine on his patients and became addicted through self-experimentation. In the latter half of the 1800s, interest in the drug became widespread, and many of cocaine's pharmacologic actions and adverse effects were elucidated during this time. In the 1880s, Koller introduced cocaine to the field of ophthalmology, and Hall introduced it to dentistry Overwiev Local anesthetics (LAs) are drugs that block the sensation of pain in the region where they are administered. LAs act by reversibly blocking the sodium channels of nerve fibers, thereby inhibiting the conduction of nerve impulses. Nerve fibers which carry pain sensation have the smallest diameter and are the first to be blocked by LAs. Loss of motor function and sensation of touch and pressure follow, depending on the duration of action and dose of the LA used. LAs can be infiltrated into skin/subcutaneous tissues to achieve local anesthesia or into the epidural/subarachnoid space to achieve regional anesthesia (e.g., spinal anesthesia, epidural anesthesia, etc.). Some LAs (lidocaine, prilocaine, tetracaine) are effective on topical application and are used before minor invasive procedures (venipuncture, bladder catheterization, endoscopy/laryngoscopy). LAs are divided into two groups based on their chemical structure. The amide group (lidocaine, prilocaine, mepivacaine, etc.) is safer and, hence, more commonly used in clinical practice. -

Prohibited Substances List
Prohibited Substances List This is the Equine Prohibited Substances List that was voted in at the FEI General Assembly in November 2009 alongside the new Equine Anti-Doping and Controlled Medication Regulations(EADCMR). Neither the List nor the EADCM Regulations are in current usage. Both come into effect on 1 January 2010. The current list of FEI prohibited substances remains in effect until 31 December 2009 and can be found at Annex II Vet Regs (11th edition) Changes in this List : Shaded row means that either removed or allowed at certain limits only SUBSTANCE ACTIVITY Banned Substances 1 Acebutolol Beta blocker 2 Acefylline Bronchodilator 3 Acemetacin NSAID 4 Acenocoumarol Anticoagulant 5 Acetanilid Analgesic/anti-pyretic 6 Acetohexamide Pancreatic stimulant 7 Acetominophen (Paracetamol) Analgesic/anti-pyretic 8 Acetophenazine Antipsychotic 9 Acetylmorphine Narcotic 10 Adinazolam Anxiolytic 11 Adiphenine Anti-spasmodic 12 Adrafinil Stimulant 13 Adrenaline Stimulant 14 Adrenochrome Haemostatic 15 Alclofenac NSAID 16 Alcuronium Muscle relaxant 17 Aldosterone Hormone 18 Alfentanil Narcotic 19 Allopurinol Xanthine oxidase inhibitor (anti-hyperuricaemia) 20 Almotriptan 5 HT agonist (anti-migraine) 21 Alphadolone acetate Neurosteriod 22 Alphaprodine Opiod analgesic 23 Alpidem Anxiolytic 24 Alprazolam Anxiolytic 25 Alprenolol Beta blocker 26 Althesin IV anaesthetic 27 Althiazide Diuretic 28 Altrenogest (in males and gelidngs) Oestrus suppression 29 Alverine Antispasmodic 30 Amantadine Dopaminergic 31 Ambenonium Cholinesterase inhibition 32 Ambucetamide Antispasmodic 33 Amethocaine Local anaesthetic 34 Amfepramone Stimulant 35 Amfetaminil Stimulant 36 Amidephrine Vasoconstrictor 37 Amiloride Diuretic 1 Prohibited Substances List This is the Equine Prohibited Substances List that was voted in at the FEI General Assembly in November 2009 alongside the new Equine Anti-Doping and Controlled Medication Regulations(EADCMR).