Multicolor Single Particle Reconstruction of Protein Complexes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Old Data and Friends Improve with Age: Advancements with the Updated Tools of Genenetwork
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.24.445383; this version posted May 25, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Old data and friends improve with age: Advancements with the updated tools of GeneNetwork Alisha Chunduri1, David G. Ashbrook2 1Department of Biotechnology, Chaitanya Bharathi Institute of Technology, Hyderabad 500075, India 2Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN 38163, USA Abstract Understanding gene-by-environment interactions is important across biology, particularly behaviour. Families of isogenic strains are excellently placed, as the same genome can be tested in multiple environments. The BXD’s recent expansion to 140 strains makes them the largest family of murine isogenic genomes, and therefore give great power to detect QTL. Indefinite reproducible genometypes can be leveraged; old data can be reanalysed with emerging tools to produce novel biological insights. To highlight the importance of reanalyses, we obtained drug- and behavioural-phenotypes from Philip et al. 2010, and reanalysed their data with new genotypes from sequencing, and new models (GEMMA and R/qtl2). We discover QTL on chromosomes 3, 5, 9, 11, and 14, not found in the original study. We narrowed down the candidate genes based on their ability to alter gene expression and/or protein function, using cis-eQTL analysis, and variants predicted to be deleterious. Co-expression analysis (‘gene friends’) and human PheWAS were used to further narrow candidates. -

The Basal Bodies of Chlamydomonas Reinhardtii Susan K
Dutcher and O’Toole Cilia (2016) 5:18 DOI 10.1186/s13630-016-0039-z Cilia REVIEW Open Access The basal bodies of Chlamydomonas reinhardtii Susan K. Dutcher1* and Eileen T. O’Toole2 Abstract The unicellular green alga, Chlamydomonas reinhardtii, is a biflagellated cell that can swim or glide. C. reinhardtii cells are amenable to genetic, biochemical, proteomic, and microscopic analysis of its basal bodies. The basal bodies contain triplet microtubules and a well-ordered transition zone. Both the mother and daughter basal bodies assemble flagella. Many of the proteins found in other basal body-containing organisms are present in the Chlamydomonas genome, and mutants in these genes affect the assembly of basal bodies. Electron microscopic analysis shows that basal body duplication is site-specific and this may be important for the proper duplication and spatial organization of these organelles. Chlamydomonas is an excellent model for the study of basal bodies as well as the transition zone. Keywords: Site-specific basal body duplication, Cartwheel, Transition zone, Centrin fibers Phylogeny and conservation of proteins Centrin, SPD2/CEP192, Asterless/CEP152; CEP70, The green lineage or Viridiplantae consists of the green delta-tubulin, and epsilon-tubulin. Chlamydomonas has algae, which include Chlamydomonas, the angiosperms homologs of all of these based on sequence conservation (the land plants), and the gymnosperms (conifers, cycads, except PLK4, CEP152, and CEP192. Several lines of evi- ginkgos). They are grouped together because they have dence suggests that CEP152, CEP192, and PLK4 interact chlorophyll a and b and lack phycobiliproteins. The green [20, 52] and their concomitant absence in several organ- algae together with the cycads and ginkgos have basal isms suggests that other mechanisms exist that allow for bodies and cilia, while the angiosperms and conifers have control of duplication [4]. -

Par6c Is at the Mother Centriole and Controls Centrosomal Protein
860 Research Article Par6c is at the mother centriole and controls centrosomal protein composition through a Par6a-dependent pathway Vale´rian Dormoy, Kati Tormanen and Christine Su¨ tterlin* Department of Developmental and Cell Biology, University of California, Irvine, Irvine, CA 92697-2300, USA *Author for correspondence ([email protected]) Accepted 3 December 2012 Journal of Cell Science 126, 860–870 ß 2013. Published by The Company of Biologists Ltd doi: 10.1242/jcs.121186 Summary The centrosome contains two centrioles that differ in age, protein composition and function. This non-membrane bound organelle is known to regulate microtubule organization in dividing cells and ciliogenesis in quiescent cells. These specific roles depend on protein appendages at the older, or mother, centriole. In this study, we identified the polarity protein partitioning defective 6 homolog gamma (Par6c) as a novel component of the mother centriole. This specific localization required the Par6c C-terminus, but was independent of intact microtubules, the dynein/dynactin complex and the components of the PAR polarity complex. Par6c depletion resulted in altered centrosomal protein composition, with the loss of a large number of proteins, including Par6a and p150Glued, from the centrosome. As a consequence, there were defects in ciliogenesis, microtubule organization and centrosome reorientation during migration. Par6c interacted with Par3 and aPKC, but these proteins were not required for the regulation of centrosomal protein composition. Par6c also associated with Par6a, which controls protein recruitment to the centrosome through p150Glued. Our study is the first to identify Par6c as a component of the mother centriole and to report a role of a mother centriole protein in the regulation of centrosomal protein composition. -
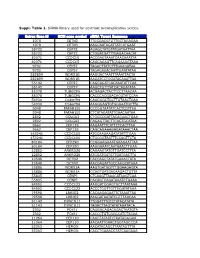
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

An Exome-Wide Sequencing Study of Lipid Response to High-Fat Meal and Fenofibrate in Caucasians from the GOLDN Cohort
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 An exome-wide sequencing study of lipid response to high-fat meal and fenofibrate in Caucasians from the GOLDN cohort Xin Geng Ping An Mary F. Feitosa Michael A. Province et al. Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Supplemental Material can be found at: http://www.jlr.org/content/suppl/2018/02/20/jlr.P080333.DC1 .html patient-oriented and epidemiological research An exome-wide sequencing study of lipid response to high-fat meal and fenofibrate in Caucasians from the GOLDN cohort Xin Geng,* Marguerite R. Irvin,† Bertha Hidalgo,† Stella Aslibekyan,† Vinodh Srinivasasainagendra,§ Ping An,‡ Alexis C. Frazier-Wood,|| Hemant K. Tiwari,§ Tushar Dave,# Kathleen Ryan,# Jose M. Ordovas,$,**,†† Robert J. Straka,§§ Mary F. Feitosa,‡ Paul N. Hopkins,‡‡ Ingrid Borecki,|| || Michael A. Province,‡ Braxton D. Mitchell,# Donna K. Arnett,1,## and Degui Zhi1,*,$$ School of Biomedical Informatics* and School of Public Health,$$ The University of Texas Health Downloaded from Science Center at Houston, Houston, TX 77030; Departments of Epidemiology† and Biostatistics,§ University of Alabama at Birmingham, Birmingham, AL 35233; Division of Statistical Genomics, Department of Genetics,‡ Washington University School of Medicine, St. Louis, MO 63110; US Department of Agriculture/Agricultural Research Service Children’s Nutrition Research Center,|| Baylor College of Medicine, Houston, TX 77030; Department of Medicine, Division -

Whole Exome Sequencing Identifies Causative Mutations in the Majority of Consanguineous Or Familial Cases with Childhood-Onset I
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Kidney Manuscript Author Int. Author manuscript; Manuscript Author available in PMC 2016 August 01. Published in final edited form as: Kidney Int. 2016 February ; 89(2): 468–475. doi:10.1038/ki.2015.317. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood- onset increased renal echogenicity Daniela A. Braun#1, Markus Schueler#1, Jan Halbritter1, Heon Yung Gee1, Jonathan D. Porath1, Jennifer A. Lawson1, Rannar Airik1, Shirlee Shril1, Susan J. Allen2, Deborah Stein1, Adila Al Kindy3, Bodo B. Beck4, Nurcan Cengiz5, Khemchand N. Moorani6, Fatih Ozaltin7,8,9, Seema Hashmi10, John A. Sayer11, Detlef Bockenhauer12, Neveen A. Soliman13,14, Edgar A. Otto2, Richard P. Lifton15,16,17, and Friedhelm Hildebrandt1,17 1Division of Nephrology, Department of Medicine, Boston Children's Hospital, Harvard Medical School, Boston, Massachusetts, USA 2Department of Pediatrics, University of Michigan, Michigan, USA 3Department of Genetics, Sultan Qaboos University Hospital, Sultanate of Oman 4Institute for Human Genetics, University of Cologne, Germany 5Baskent University, School of Medicine, Adana Medical Training and Research Center, Department of Pediatric Nephrology, Adana, Turkey 6Department of Pediatric Nephrology, National Institute of Child Health, Karachi 75510, Pakistan 7Faculty of Medicine, Department of Pediatric Nephrology, Hacettepe University, Ankara, Turkey 8Nephrogenetics Laboratory, Faculty of Medicine, -

TALPID3 and ANKRD26 Selectively Orchestrate FBF1 Localization and Cilia Gating
ARTICLE https://doi.org/10.1038/s41467-020-16042-w OPEN TALPID3 and ANKRD26 selectively orchestrate FBF1 localization and cilia gating Hao Yan 1,2,3,7, Chuan Chen4,7, Huicheng Chen1,2,3, Hui Hong 1, Yan Huang4,5, Kun Ling4,5, ✉ ✉ Jinghua Hu4,5,6 & Qing Wei 3 Transition fibers (TFs) regulate cilia gating and make the primary cilium a distinct functional entity. However, molecular insights into the biogenesis of a functional cilia gate remain 1234567890():,; elusive. In a forward genetic screen in Caenorhabditis elegans, we uncover that TALP-3, a homolog of the Joubert syndrome protein TALPID3, is a TF-associated component. Genetic analysis reveals that TALP-3 coordinates with ANKR-26, the homolog of ANKRD26, to orchestrate proper cilia gating. Mechanistically, TALP-3 and ANKR-26 form a complex with key gating component DYF-19, the homolog of FBF1. Co-depletion of TALP-3 and ANKR-26 specifically impairs the recruitment of DYF-19 to TFs. Interestingly, in mammalian cells, TALPID3 and ANKRD26 also play a conserved role in coordinating the recruitment of FBF1 to TFs. We thus report a conserved protein module that specifically regulates the functional component of the ciliary gate and suggest a correlation between defective gating and cilio- pathy pathogenesis. 1 CAS Key Laboratory of Insect Developmental and Evolutionary Biology, CAS Center for Excellence in Molecular Plant Sciences, Institute of Plant Physiology and Ecology, Chinese Academy of Sciences, Shanghai 200032, China. 2 University of Chinese Academy of Sciences, Beijing 100039, China. 3 Center for Reproduction and Health Development, Institute of Biomedicine and Biotechnology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences (CAS), Shenzhen 518055, China. -

CEP164-Null Cells Generated by Genome Editing Show a Ciliation Defect with Intact DNA Repair Capacity Owen M
© 2016. Published by The Company of Biologists Ltd | Journal of Cell Science (2016) 129, 1769-1774 doi:10.1242/jcs.186221 SHORT REPORT CEP164-null cells generated by genome editing show a ciliation defect with intact DNA repair capacity Owen M. Daly1, David Gaboriau1,*, Kadin Karakaya2, Sinéad King1, Tiago J. Dantas1,‡, Pierce Lalor3, Peter Dockery3, Alwin Krämer2 and Ciaran G. Morrison1,§ ABSTRACT induced by the removal of growth factors, facilitates ciliogenesis Primary cilia are microtubule structures that extend from the distal end (Kobayashi and Dynlacht, 2011). Current models associate primary of the mature, mother centriole. CEP164 is a component of the distal cilia with cell cycle exit and reduced proliferation, although the appendages carried by the mother centriole that is required for underlying mechanisms of such a link are not well defined (Goto primary cilium formation. Recent data have implicated CEP164 as a et al., 2013). CEP164 ciliopathy gene and suggest that CEP164 plays some roles in the encodes a centriolar appendage protein that is required DNA damage response (DDR). We used reverse genetics to test the for ciliogenesis (Graser et al., 2007; Schmidt et al., 2012). It has also role of CEP164 in the DDR. We found that conditional depletion of been implicated in modulating the DNA damage response (DDR), CEP164 in chicken DT40 cells using an auxin-inducible degron led to particularly CHK1 (Sivasubramaniam et al., 2008). CEP164 was no increase in sensitivity to DNA damage induced by ionising or initially identified in a proteomic analysis of the centrosome and, ultraviolet irradiation. Disruption of CEP164 in human retinal later, as a component of the distal appendages whose depletion by pigmented epithelial cells blocked primary cilium formation but did small interfering RNA (siRNA) treatment caused a marked not affect cellular proliferation or cellular responses to ionising or reduction in primary cilium formation (Andersen et al., 2003; ultraviolet irradiation. -

Cep164 Mediates Vesicular Docking to the Mother Centriole During Early Steps of Ciliogenesis
JCB: Article Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis Kerstin N. Schmidt,1 Stefanie Kuhns,1 Annett Neuner,2 Birgit Hub,3 Hanswalter Zentgraf,3 and Gislene Pereira1 1DKFZ-ZMBH Alliance, German Cancer Research Center, 69120 Heidelberg, Germany 2DKFZ-ZMBH Alliance, Center for Molecular and Cellular Biology, 69120 Heidelberg, Germany 3Department of Tumor Virology, German Cancer Research Center, 69120 Heidelberg, Germany ilia formation is a multi-step process that starts We show that Cep164 was indispensable for the docking with the docking of a vesicle at the distal part of of vesicles at the mother centriole. Using biochemical and C the mother centriole. This step marks the conver- functional assays, we identified the components of the sion of the mother centriole into the basal body, from vesicular transport machinery, the GEF Rabin8 and the which axonemal microtubules extend to form the ciliary GTPase Rab8, as interacting partners of Cep164. We compartment. How vesicles are stably attached to the propose that Cep164 is targeted to the apical domain of mother centriole to initiate ciliary membrane biogenesis the mother centriole to provide the molecular link between is unknown. Here, we investigate the molecular role of the mother centriole and the membrane biogenesis ma- the mother centriolar component Cep164 in ciliogenesis. chinery that initiates cilia formation. Introduction Primary cilia are evolutionarily conserved organelles that play promotes cilia formation and extension via anterograde and retro- an essential role in embryonic development and tissue homeo- grade transport of cargo along the ciliary microtubules (Pedersen stasis in adulthood (D’Angelo and Franco, 2009; Tasouri and and Rosenbaum, 2008). -

ARL13B, PDE6D, and CEP164 Form a Functional Network for INPP5E Ciliary Targeting
ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting Melissa C. Humberta,b, Katie Weihbrechta,b, Charles C. Searbyb,c, Yalan Lid, Robert M. Poped, Val C. Sheffieldb,c, and Seongjin Seoa,1 aDepartment of Ophthalmology and Visual Sciences, bDepartment of Pediatrics, cHoward Hughes Medical Institute, and dProteomics Facility, University of Iowa, Iowa City, IA 52242 Edited by Kathryn V. Anderson, Sloan-Kettering Institute, New York, NY, and approved October 19, 2012 (received for review June 28, 2012) Mutations affecting ciliary components cause a series of related polydactyly, skeletal defects, cleft palate, and cerebral develop- genetic disorders in humans, including nephronophthisis (NPHP), mental defects (11). Inactivation of Inpp5e in adult mice results in Joubert syndrome (JBTS), Meckel-Gruber syndrome (MKS), and Bar- obesity and photoreceptor degeneration. Interestingly, many pro- det-Biedl syndrome (BBS), which are collectively termed “ciliopa- teins that localize to cilia, including INPP5E, RPGR, PDE6 α and thies.” Recent protein–protein interaction studies combined with β subunits, GRK1 (Rhodopsin kinase), and GNGT1 (Transducin γ genetic analyses revealed that ciliopathy-related proteins form sev- chain), are prenylated (either farnesylated or geranylgeranylated), eral functional networks/modules that build and maintain the pri- and mutations in these genes or genes involved in their prenylation mary cilium. However, the precise function of many ciliopathy- (e.g., AIPL1 and RCE1) lead to photoreceptor -

Ciliary Genes in Renal Cystic Diseases
cells Review Ciliary Genes in Renal Cystic Diseases Anna Adamiok-Ostrowska * and Agnieszka Piekiełko-Witkowska * Department of Biochemistry and Molecular Biology, Centre of Postgraduate Medical Education, 01-813 Warsaw, Poland * Correspondence: [email protected] (A.A.-O.); [email protected] (A.P.-W.); Tel.: +48-22-569-3810 (A.P.-W.) Received: 3 March 2020; Accepted: 5 April 2020; Published: 8 April 2020 Abstract: Cilia are microtubule-based organelles, protruding from the apical cell surface and anchoring to the cytoskeleton. Primary (nonmotile) cilia of the kidney act as mechanosensors of nephron cells, responding to fluid movements by triggering signal transduction. The impaired functioning of primary cilia leads to formation of cysts which in turn contribute to development of diverse renal diseases, including kidney ciliopathies and renal cancer. Here, we review current knowledge on the role of ciliary genes in kidney ciliopathies and renal cell carcinoma (RCC). Special focus is given on the impact of mutations and altered expression of ciliary genes (e.g., encoding polycystins, nephrocystins, Bardet-Biedl syndrome (BBS) proteins, ALS1, Oral-facial-digital syndrome 1 (OFD1) and others) in polycystic kidney disease and nephronophthisis, as well as rare genetic disorders, including syndromes of Joubert, Meckel-Gruber, Bardet-Biedl, Senior-Loken, Alström, Orofaciodigital syndrome type I and cranioectodermal dysplasia. We also show that RCC and classic kidney ciliopathies share commonly disturbed genes affecting cilia function, including VHL (von Hippel-Lindau tumor suppressor), PKD1 (polycystin 1, transient receptor potential channel interacting) and PKD2 (polycystin 2, transient receptor potential cation channel). Finally, we discuss the significance of ciliary genes as diagnostic and prognostic markers, as well as therapeutic targets in ciliopathies and cancer. -

Tethering of an E3 Ligase by PCM1 Regulates the Abundance Of
RESEARCH ARTICLE Tethering of an E3 ligase by PCM1 regulates the abundance of centrosomal KIAA0586/Talpid3 and promotes ciliogenesis Lei Wang†, Kwanwoo Lee†, Ryan Malonis, Irma Sanchez, Brian D Dynlacht* Department of Pathology, New York University Cancer Institute, New York University School of Medicine, New York, United States Abstract To elucidate the role of centriolar satellites in ciliogenesis, we deleted the gene encoding the PCM1 protein, an integral component of satellites. PCM1 null human cells show marked defects in ciliogenesis, precipitated by the loss of specific proteins from satellites and their relocation to centrioles. We find that an amino-terminal domain of PCM1 can restore ciliogenesis and satellite localization of certain proteins, but not others, pinpointing unique roles for PCM1 and a group of satellite proteins in cilium assembly. Remarkably, we find that PCM1 is essential for tethering the E3 ligase, Mindbomb1 (Mib1), to satellites. In the absence of PCM1, Mib1 destabilizes Talpid3 through poly-ubiquitylation and suppresses cilium assembly. Loss of PCM1 blocks ciliogenesis by abrogating recruitment of ciliary vesicles associated with the Talpid3-binding protein, Rab8, which can be reversed by inactivating Mib1. Thus, PCM1 promotes ciliogenesis by tethering a key E3 ligase to satellites and restricting it from centrioles. DOI: 10.7554/eLife.12950.001 *For correspondence: Brian. [email protected] †These authors contributed Introduction equally to this work Primary cilia are antenna-like organelles that protrude from the surface of many differentiated cells Competing interests: The and serve to orchestrate key signaling events required for development (reviewed in Kobayashi and authors declare that no Dynlacht, 2011).