Primepcr™Assay Validation Report
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Title of the Article
Mechanism-Anchored Profiling Derived from Epigenetic Networks Predicts Outcome in Acute Lymphoblastic Leukemia Xinan Yang, PhD1, Yong Huang, MD1, James L Chen, MD1, Jianming Xie, MSc2, Xiao Sun, PhD2, Yves A Lussier, MD1,3,4§ 1Center for Biomedical Informatics and Section of Genetic Medicine, Department of Medicine, The University of Chicago, Chicago, IL 60637 USA 2State Key Laboratory of Bioelectronics, Southeast University, 210096 Nanjing, P.R.China 3The University of Chicago Cancer Research Center, and The Ludwig Center for Metastasis Research, The University of Chicago, Chicago, IL 60637 USA 4The Institute for Genomics and Systems Biology, and the Computational Institute, The University of Chicago, Chicago, IL 60637 USA §Corresponding author Email addresses: XY: [email protected] YH: [email protected] JC: [email protected] JX: [email protected] XS: [email protected] YL: [email protected] - 1 - Abstract Background Current outcome predictors based on “molecular profiling” rely on gene lists selected without consideration for their molecular mechanisms. This study was designed to demonstrate that we could learn about genes related to a specific mechanism and further use this knowledge to predict outcome in patients – a paradigm shift towards accurate “mechanism-anchored profiling”. We propose a novel algorithm, PGnet, which predicts a tripartite mechanism-anchored network associated to epigenetic regulation consisting of phenotypes, genes and mechanisms. Genes termed as GEMs in this network meet all of the following criteria: (i) they are co-expressed with genes known to be involved in the biological mechanism of interest, (ii) they are also differentially expressed between distinct phenotypes relevant to the study, and (iii) as a biomodule, genes correlate with both the mechanism and the phenotype. -

Supp Data.Pdf
Supplementary Methods Verhaak sub-classification The Verhaak called tumor subtype was assigned as determined in the Verhaak et al. manuscript, for those TCGA tumors that were included at the time. TCGA data for expression (Affymetrix platform, level 1) and methylation (Infinium platform level 3) were downloaded from the TCGA portal on 09/29/2011. Affymetrix data were processed using R and Biocondutor with a RMA algorithm using quantile normalization and custom CDF. Level 3 methylation data has already been processed and converted to a beta-value. The genes that make up the signature for each of the 4 Verhaak groups (i.e. Proneural, Neural, Mesenchymal, and Classical) were downloaded. For each tumor, the averaged expression of the 4 genes signatures was calculated generating 4 ‘metagene’ scores, one for each subtype, (Colman et al.2010) for every tumor. Then each subtype metagene score was z-score corrected to allow comparison among the 4 metagenes. Finally, for each tumor the subtype with the highest metagene z-score among the four was used to assign subtype. Thus by this method, a tumor is assigned to the group to which it has the strongest expression signature. The shortcoming is that if a tumor has a ‘dual personality’ – for instance has strong expression of both classical and mesenchymal signature genes, there is some arbitrariness to the tumor’s assignment. Western Blot Analysis and cell fractionation Western blot analysis was performed using standard protocols. To determine protein expression we used the following antibodies: TAZ (Sigma and BD Biosciences), YAP (Santa Cruz Biotechnology), CD44 (Cell Signaling), FN1 (BD Biosciences), TEAD1, TEAD2, TEAD3, TEAD4 (Santa Cruz Biotechnology), MST1, p-MST1, MOB1, LATS1, LATS2, 14-3-3-, 1 ACTG2, (Cell Signaling), p-LATS1/2 (Abcam), Flag (Sigma Aldrich), Actin (Calbiochem), CAV2 (BD Biosciences), CTGF (Santa Cruz), RUNX2 (Sigma Aldrich), Cylin A, Cyclin E, Cyclin B1, p-cdk1, p-cdk4 (Cell Signaling Technologies). -

Canonical WNT Signalling Determines Lineage Specificity in Wilms Tumour
Oncogene (2009) 28, 1063–1075 & 2009 Macmillan Publishers Limited All rights reserved 0950-9232/09 $32.00 www.nature.com/onc ORIGINAL ARTICLE Canonical WNT signalling determines lineage specificity in Wilms tumour R Fukuzawa, MR Anaka, RJ Weeks, IM Morison and AE Reeve Cancer Genetics Laboratory, Department of Biochemistry, University of Otago, Dunedin, New Zealand Wilms tumours (WTs) have two distinct types of histology to translocation of the protein to the nucleus (Clevers, with or without ectopic mesenchymal elements, suggesting 2006). Nuclear accumulation of b-catenin is therefore a that WTs arise from either the mesenchymal or epithelial surrogate marker for activation of the WNT-signalling nephrogenic lineages. Regardless of the presence or pathway in many types of cancers (Clevers, 2006). absence of CTNNB1 mutations, nuclear accumulation of Wilms tumour (WT) is an embryonal tumour that b-catenin is often observed in WTs with ectopic mesench- recapitulates kidney development. WT typically has a ymal elements. Here, we addressed the relationship triphasic histology, which consists of varying propor- between the WNT-signalling pathway and lineage in tions of blastemal, epithelial and stromal elements WTs by examining CTNNB1 and WT1 mutations, (Beckwith et al., 1990). The stromal tissues occasionally nuclear accumulation of b-catenin, tumour histology and contain ectopic mesenchymal elements such as rhabdo- gene expression profiles. In addition, we screened for myoblasts, chondrocytes and adipocytes. Remarkably, mutations in WTX, which has been proposed to be a the presence or absence of ectopic mesenchymal negative regulator of the canonical WNT-signalling elements in WT histology is associated with specific pathway. Unsupervised clustering analysis identified two precursor lesions, intralobar and perilobar nephrogenic classes of tumours: mesenchymal lineage WNT-dependent rests (ILNR and PLNR), respectively (Beckwith et al., tumours, and epithelial lineage WNT-independent tu- 1990). -

Theses Digitisation: This Is a Digitised
https://theses.gla.ac.uk/ Theses Digitisation: https://www.gla.ac.uk/myglasgow/research/enlighten/theses/digitisation/ This is a digitised version of the original print thesis. Copyright and moral rights for this work are retained by the author A copy can be downloaded for personal non-commercial research or study, without prior permission or charge This work cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given Enlighten: Theses https://theses.gla.ac.uk/ [email protected] Identification ofTopBPl Chromatin Modification Domains and Transcriptional Targets Roni H G Wright A thesis submitted to the University o f Glasgow, Faculty of Veterinary Medicine for the degree o f Doctor of Philosophy UNIVERSITY of GLASGOW i r % Division of Pathological Sciences Faculty of Veterinary Medicine September 2007 © Roni Wright :v & ProQuest Number: 10391063 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 10391063 Published by ProQuest LLO (2017). C o pyright of the Dissertation is held by the Author. -
![Downloaded from NCBI Website [44] and Installed Extension Assay to a Local Server](https://docslib.b-cdn.net/cover/8559/downloaded-from-ncbi-website-44-and-installed-extension-assay-to-a-local-server-2748559.webp)
Downloaded from NCBI Website [44] and Installed Extension Assay to a Local Server
BMC Genomics BioMed Central Methodology article Open Access A highly sensitive and specific system for large-scale gene expression profiling Guohong Hu†, Qifeng Yang†, Xiangfeng Cui, Gang Yue, Marco A Azaro, Hui- Yun Wang and Honghua Li* Address: Department of Molecular Genetics, Microbiology and Immunology/The Cancer Institute of New Jersey, University of Medicine and Dentistry of New Jersey Robert Wood Johnson Medical School, Piscataway, New Jersey 08854, USA Email: Guohong Hu - [email protected]; Qifeng Yang - [email protected]; Xiangfeng Cui - [email protected]; Gang Yue - [email protected]; Marco A Azaro - [email protected]; Hui-Yun Wang - [email protected]; Honghua Li* - [email protected] * Corresponding author †Equal contributors Published: 10 January 2008 Received: 18 January 2007 Accepted: 10 January 2008 BMC Genomics 2008, 9:9 doi:10.1186/1471-2164-9-9 This article is available from: http://www.biomedcentral.com/1471-2164/9/9 © 2008 Hu et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Rapid progress in the field of gene expression-based molecular network integration has generated strong demand on enhancing the sensitivity and data accuracy of experimental systems. To meet the need, a high-throughput gene profiling system of high specificity and sensitivity has been developed. Results: By using specially designed primers, the new system amplifies sequences in neighboring exons separated by big introns so that mRNA sequences may be effectively discriminated from other highly related sequences including their genes, unprocessed transcripts, pseudogenes and pseudogene transcripts. -

A Second Generation Human Haplotype Map of Over 3.1 Million Snps the International Hapmap Consortium1
doi: 10.1038/nature06258 SUPPLEMENTARY INFORMATION A second generation human haplotype map of over 3.1 million SNPs The International HapMap Consortium1 Supplementary material S1 The density of common SNPs in the Phase II HapMap and the assembled human genome S2 Analysis of data quality S2.1 Analysis of amplicon structure to genotyping error S2.2 Analysis of genotype discordance from overlap with Seattle SNPs S2.3 Analysis of genotype discordance from fosmid end sequences S2.4 Analysis of monomorphism/polymorphism discrepancies S2.5 Interchromosomal LD S3. Analysis of population stratification S4. Analysis of relatedness S5. Segmental analysis of relatedness S6. Analysis of homozygosity S7. Perlegen genotyping protocols Supplementary tables Legends to supplementary figures 1 See end of manuscript for Consortium details www.nature.com/nature 1 doi: 10.1038/nature06258 SUPPLEMENTARY INFORMATION Supplementary text 1. The density of common SNPs in the Phase II HapMap and the assembled human genome. To estimate the fraction of all common variants on the autosomes that have been successfully genotyped in the consensus Phase II HapMap we note that in YRI (release 21) there are 2,334,980 SNPs with MAF≥0.05. Across the autosomes, the completed reference sequence assembled in contigs is 2.68 billion bp. Assuming that the allele frequency distribution in the YRI is well approximated by that of a simple coalescent model and using an estimate of the population mutation rate of θ = 1.2 per kb for African populations1,2 the expected number of variants with MAF≥0.05 in a sample of 120 chromosomes is 114 = θ E(S MAF≥5% ) L ∑ /1 i i=6 where L is the total length of the sequence3. -
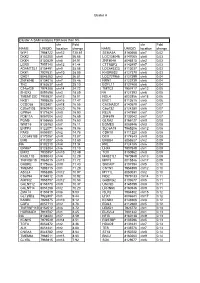
Copy of Supplementary Table 2
Cluster A Cluster A SAM analysis FDR less than 5% chr Fold chr Fold NAME UNIQID location change NAME UNIQID location change WIF1 7964722 chr12 130.61 SEMA3A 8140668 chr7 0.02 DKK2 8102200 chr4 56.66 LOC138046 8147065 chr8 0.03 DKK4 8150529 chr8 54.91 ZNF804A 8046815 chr2 0.03 LGR5 7957140 chr12 41.44 CTTNBP2 8142497 chr7 0.03 ADAMTSL1 8154491 chr9 35.08 LOC645323 8113037 chr5 0.03 DKK1 7927631 chr10 28.09 KHDRBS2 8127370 chr6 0.03 GAD1 8046283 chr2 26.81 LOC727966 8127399 chr6 0.04 ZNF804B 8134018 chr7 25.46 NRN1 8123739 chr6 0.04 TNC 8163637 chr9 25.10 EGFL11 8127408 chr6 0.04 C14orf29 7974288 chr14 24.72 TMTC2 7957417 chr12 0.05 SHOX2 8091698 chr3 18.89 NA 8127393 chr6 0.05 TMEM132C 7959827 chr12 18.01 NOL4 8022856 chr18 0.06 NKD1 7995525 chr16 17.47 ENC1 8112615 chr5 0.06 CCDC68 8023401 chr18 16.55 CACNA2D1 8140579 chr7 0.07 C20orf103 8060940 chr20 16.06 C6orf32 8124280 chr6 0.07 GPR64 8171624 chrX 15.90 RELN 8141950 chr7 0.07 PDE11A 8057004 chr2 15.69 ZNF679 8133042 chr7 0.07 PGM5 8155665 chr9 15.60 GLRA2 8166127 chrX 0.08 WNT16 8135763 chr7 15.11 EOMES 8085946 chr3 0.08 ENPP3 8122071 chr6 15.06 SLC6A15 7965206 chr12 0.08 PAX3 8059301 chr2 14.78 CDH18 8111220 chr5 0.08 LOC645188 8170257 chrX 13.87 DCX 8174543 chrX 0.08 EMB 8112007 chr5 13.60 ERBB4 8058627 chr2 0.09 NA 8102210 chr4 13.34 PRL 8124185 chr6 0.09 EPHA7 8128284 chr6 13.10 LHX4 7907849 chr1 0.09 EMX2 7930857 chr10 12.49 TOX 8150962 chr8 0.09 RASL11B 8095043 chr4 12.29 MAB21L1 7970949 chr13 0.09 TNFRSF19 7968015 chr13 11.72 MPP3 8015846 chr17 0.09 GABRE 8175666 chrX 11.40 -

Supplementary Data
SUPPLEMENTARY METHODS 1) Characterisation of OCCC cell line gene expression profiles using Prediction Analysis for Microarrays (PAM) The ovarian cancer dataset from Hendrix et al (25) was used to predict the phenotypes of the cell lines used in this study. Hendrix et al (25) analysed a series of 103 ovarian samples using the Affymetrix U133A array platform (GEO: GSE6008). This dataset comprises clear cell (n=8), endometrioid (n=37), mucinous (n=13) and serous epithelial (n=41) primary ovarian carcinomas and samples from 4 normal ovaries. To build the predictor, the Prediction Analysis of Microarrays (PAM) package in R environment was employed (http://rss.acs.unt.edu/Rdoc/library/pamr/html/00Index.html). When more than one probe described the expression of a given gene, we used the probe with the highest median absolute deviation across the samples. The dataset from Hendrix et al. (25) and the dataset of OCCC cell lines described in this manuscript were then overlaid on the basis of 11536 common unique HGNC gene symbols. Only the 99 primary ovarian cancers samples and the four normal ovary samples were used to build the predictor. Following leave one out cross-validation, a predictor based upon 126 genes was able to identify correctly the four distinct phenotypes of primary ovarian tumour samples with a misclassification rate of 18.3%. This predictor was subsequently applied to the expression data from the 12 OCCC cell lines to determine the likeliest phenotype of the OCCC cell lines compared to primary ovarian cancers. Posterior probabilities were estimated for each cell line in comparison to the following phenotypes: clear cell, endometrioid, mucinous and serous epithelial. -

X Chromosome Cdna Microarray Screening Identifies a Functional PLP2 Promoter Polymorphism Enriched in Patients with X-Linked Mental Retardation
Downloaded from genome.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press Methods X chromosome cDNA microarray screening identifies a functional PLP2 promoter polymorphism enriched in patients with X-linked mental retardation Lilei Zhang,1 Chunfa Jie,2 Cassandra Obie,1 Fatima Abidi,3 Charles E. Schwartz,3 Roger E. Stevenson,3 David Valle,1 and Tao Wang1,4 1McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore 21205, Maryland; 2Microarray Core Facility, Johns Hopkins University School of Medicine, Baltimore , Maryland; 3Greenwood Genetic Center, Greenwood 29646, South Carolina X-linked Mental Retardation (XLMR) occurs in 1 in 600 males and is highly genetically heterogeneous. We used a novel human X chromosome cDNA microarray (XCA) to survey the expression profile of X-linked genes in lymphoblasts of XLMR males. Genes with altered expression verified by Northern blot and/or quantitative PCR were considered candidates. To validate this approach, we documented the expected changes of expression in samples from a patient with a known X chromosome microdeletion and from patients with multiple copies of the X chromosome. We used our XCA to survey lymphoblast RNA samples from 43 unrelated XLMR males and found 15 genes with significant (Ն1.5-fold) reduction in expression in at least one proband. Of these, subsequent analysis confirmed altered expression in 12. We followed up one, PLP2, at Xp11.23, which exhibits approximately fourfold decreased expression in two patients. Sequencing analysis in both patients revealed a promoter variant, −113C>A, that alters the core-binding site of the transcription factor ELK1. -

Supplementary Tables
Supplementary Tables Supplementary Table 1. Differentially methylated genes in correlation with their expression pattern in the A4 progression model A. Hypomethylated–upregulated Genes (n= 76) ALOX5 RRAD RTN4R DSCR6 FGFR3 HTR7 WNT3A POGK PLCD3 ALPPL2 RTEL1 SEMA3B DUSP5 FOSB ITGB4 MEST PPL PSMB8 ARHGEF4 BST2 SEMA7A SLC12A7 FOXQ1 KCTD12 LETM2 PRPH PXMP2 ARNTL2 CDH3 SHC2 SLC20A2 HSPA2 KIAA0182 LIMK2 NAB1 RASIP1 ASRGL1 CLDN3 DCBLD1 SNX10 SSH1 KREMEN2 LIPE NDRG2 ATF3 CLU DCHS1 SOD3 ST3GAL4 MAL LRRC1 NR3C2 ATP8B3 CYC1 DGCR8 EBAG9 SYNGR1 TYMS MCM2 NRG2 RHOF DAGLA DISP2 FAM19A5 TNNI3 UNC5B MYB PAK6 RIPK4 DAZAP1 DOCK3 FBXO6 HSPA4L WHSC1 PNMT PCDH1 B. Hypermethylated-downregulated Genes (n= 31) ARHGAP22 TNFSF9 KLF6 LRP8 NRP1 PAPSS2 SLC43A2 TBC1D16 ASB2 DZIP1 TPM1 MDGA1 NRP2 PIK3CD SMARCA2 TLL2 C18orf1 FBN1 LHFPL2 TRIO NTNG2 PTGIS SOCS2 TNFAIP8 DIXDC1 KIFC3 LMO1 NR3C1 ODZ3 PTPRM SYNPO Supplementary Table 2. Genes enriched for different histone methylation marks in A4 progression model identified through ChIP-on-chip a. H3K4me3 (n= 978) AATF C20orf149 CUL3 FOXP1 KATNA1 NEGR1 RAN SPIN2B ABCA7 C20orf52 CWF19L1 FRK KBTBD10 NEIL1 RANBP2 SPPL2A ABCC9 C21orf13-SH3BGR CXCL3 FSIP1 KBTBD6 NELF RAPGEF3 SPRY4 ABCG2 C21orf45 CYC1 FUK KCMF1 NFKB2 RARB SPRYD3 ABHD7 C22orf32 CYorf15A FXR2 KCNH7 NGDN RASAL2 SPTLC2 ACA15 C2orf18 DAXX FZD9 KCNMB4 NKAP RASD1 SRFBP1 ACA26 C2orf29 DAZ3 G6PD KCTD18 NKTR RASEF SRI ACA3 C2orf32 DBF4 GABPB2 KDELR2 NNT RASGRF1 SRM ACA48 C2orf55 DBF4B GABRA5 KIAA0100 NOL5A RASSF1 SSH2 ACAT1 C3orf44 DBI GADD45B KIAA0226 NOLC1 RASSF3 SSH3 ACSL5 -

Epigenetic Silencing Mediated Through Activated PI3K/AKT Signaling in Breast Cancer
SUPPLEMENTARY: Epigenetic Silencing Mediated Through Activated PI3K/AKT Signaling in Breast Cancer Tao Zuo, Ta-Ming Liu, Xun Lan, Yu-I Weng, Rulong Shen, Fei Gu, Yi-Wen Huang, Sandya Liyanarachchi, Daniel E. Deatherage, Pei-Yin Hsu, Cenny Taslim, Bhuvaneswari Ramaswamy, Charles L. Shapiro, Huey-Jen L. Lin, Alfred S. L. Cheng, Victor X. Jin, and Tim H.-M. Huang 1 Table of Content: 1. Fig. S1. Elevated AKT1 kinase activity in myrAKT1- transfected MCF10A cells. ---------------------------------------------------------------------------------------------------- page 4 2. Fig. S2. Genome-wide view of H3K27me3 in mammary epithelial cells. ---------------------------------------------------------------------------------------------------- page 5 3. Fig. S3. Biological functions of PI3K/AKT- downstream genes. ----------------- page 6 4. Fig. S4. Genomic maps of 40 genes. ------------------------------------------------- page 8 5. Fig. S5. Comparing DNA methylation levels of four CpG islands measured by meDIP-qPCR and MassARRAY.------------------------------------------------------- page 11 6. Fig. S6. Validation of DNA methylation by MBDCap-seq.----------------------- page 13 7. Table S1. Summary of H3K27me3 ChIP-seq data ------------------------------ page 14 8. Table S2. Distribution of H3K27me3 in MCF10A genome ---------------------- page 14 9. Table S3. Genes (n=488) enriched with H3K27me3 and downregulated by PI3K/AKT- signaling ---------------------------------------------------------------------- page 15 10. Table S4. Genes (n=1005) enriched -

Gene Ontology Analysis of Arthrogryposis (Multiple Congenital Contractures)
Received: 5 March 2019 Revised: 13 June 2019 Accepted: 17 July 2019 DOI: 10.1002/ajmg.c.31733 RESEARCH ARTICLE Gene ontology analysis of arthrogryposis (multiple congenital contractures) Jeff Kiefer1 | Judith G. Hall2,3 1Systems Oncology, Scottsdale, Arizona Abstract 2Department of Medical Genetics, University of British Columbia and BC Children's In 2016, we published an article applying Gene Ontology Analysis to the genes that had Hospital, Vancouver, British Columbia, Canada been reported to be associated with arthrogryposis (multiple congenital contractures) (Hall 3Department of Pediatrics, University of & Kiefer, 2016). At that time, 320 genes had been reported to have mutations associated British Columbia and BC Children's Hospital, Vancouver, British Columbia, Canada with arthrogryposis. All were associated with decreased fetal movement. These 320 genes were analyzed by biological process and cellular component categories, and yielded 22 Correspondence Judith G. Hall, Department of Medical distinct groupings. Since that time, another 82 additional genes have been reported, now Genetics, BC Children's Hospital, 4500 Oak totaling 402 genes, which when mutated, are associated with arthrogryposis (arthrogryposis Street, Room C234, Vancouver, British Columbia V6H 3N1, Canada. multiplex congenita). So, we decided to update the analysis in order to stimulate further Email: [email protected] research and possible treatment. Now, 29 groupings can be identified, but only 19 groups have more than one gene. KEYWORDS arthrogryposis, developmental pathways, enrichment analysis, gene ontology, multiple congenital contractures 1 | INTRODUCTION polyhydramnios, decreased gut mobility and shortened gut, short umbili- cal cord, skin changes, and multiple joints with limitation of movement, Arthrogryposis is the term that has been used for the last century including limbs, jaw, and spine).