Myo-Inositol Supplementation Augments Visual System Maturation in Mice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Abstract Book
Table of Contents Tuesday, May 25, 2021 ............................................................................................................................................................... 5 T1. Astrocyte-Specific Expression of the Extracellular Matrix Gene HtrA1 Regulates Susceptibility to Stress in a Sex- Specific Manner ....................................................................................................................................................................... 5 T2. Plexin-B2 Regulates Migratory Plasticity of Glioblastoma Cells in a 3D-Printed Micropattern Device ............................. 5 T3. Pathoanatomical Mapping of Differential MAPT Expression and Splicing in Progressive Supranuclear Palsy ............... 5 T4. Behavioral Variability in Response to Chronic Stress and Morphine in BXD and Parental Mouse Lines......................... 6 T5. Thyroid-Stimulating Hormone Receptor Regulates Anxiety .............................................................................................. 6 T6. Drugs That Inhibit Microglial Inflammation Also Ameliorate Aβ1-42 Induced Toxicity in C. Elegans ............................... 7 T7. Phosphodiesterase 1b is an Upstream Regulator of a Key Gene Network in the Nucleus Accumbens Driving Addiction- Like Behaviors ......................................................................................................................................................................... 7 T8. Reduced Gap Effect in Children With FOXP1 Syndrome and Autism Spectrum -

Astroglial Networks Scale Synaptic Activity and Plasticity
Astroglial networks scale synaptic activity and plasticity Ulrike Pannascha, Lydia Vargováb,c, Jürgen Reingruberd, Pascal Ezana, David Holcmand, Christian Giaumea, Eva Sykováb,c, and Nathalie Rouacha,1 aCenter for Interdisciplinary Research in Biology, Centre National de la Recherche Scientifique Unité Mixte de Recherche 7241/Institut National de la Santé et de la Recherche Médicale U1050, Collège de France, 75005 Paris, France; bDepartment of Neuroscience and Center for Cell therapy and Tissue Repair, Charles University, Second Faculty of Medicine, 150 06 Prague, Czech Republic; cDepartment of Neuroscience, Institute of Experimental Medicine of the Academy of Sciences of the Czech Republic, 142 20 Prague, Czech Republic; and dInstitut de Biologie de l’Ecole Normale Supérieure, Centre National de la Recherche Scientifique Unité Mixte de Recherche 8197/Institut National de la Santé et de la Recherche Médicale U1024, Department of Computational Biology and Mathematics, Ecole Normale Supérieure, Paris, 75005, France Edited* by Roger A. Nicoll, University of California, San Francisco, CA, and approved April 11, 2011 (received for review November 22, 2010) Astrocytes dynamically interact with neurons to regulate synaptic during synaptic activity. This impairment results in increased transmission. Although the gap junction proteins connexin 30 neuronal excitability, release probability, glutamate spillover, and (Cx30) and connexin 43 (Cx43) mediate the extensive network insertion of postsynaptic AMPARs, unsilencing synapses. Alto- organization of astrocytes, their role in synaptic physiology is un- gether, our findings indicate that gap junction-mediated astroglial known. Here we show, by inactivating Cx30 and Cx43 genes, that networks control synaptic strength by modulating extracellular astroglial networks tone down hippocampal synaptic transmission homeostasis. -

Specific Labeling of Synaptic Schwann Cells Reveals Unique Cellular And
RESEARCH ARTICLE Specific labeling of synaptic schwann cells reveals unique cellular and molecular features Ryan Castro1,2,3, Thomas Taetzsch1,2, Sydney K Vaughan1,2, Kerilyn Godbe4, John Chappell4, Robert E Settlage5, Gregorio Valdez1,2,6* 1Department of Molecular Biology, Cellular Biology, and Biochemistry, Brown University, Providence, United States; 2Center for Translational Neuroscience, Robert J. and Nancy D. Carney Institute for Brain Science and Brown Institute for Translational Science, Brown University, Providence, United States; 3Neuroscience Graduate Program, Brown University, Providence, United States; 4Fralin Biomedical Research Institute at Virginia Tech Carilion, Roanoke, United States; 5Department of Advanced Research Computing, Virginia Tech, Blacksburg, United States; 6Department of Neurology, Warren Alpert Medical School of Brown University, Providence, United States Abstract Perisynaptic Schwann cells (PSCs) are specialized, non-myelinating, synaptic glia of the neuromuscular junction (NMJ), that participate in synapse development, function, maintenance, and repair. The study of PSCs has relied on an anatomy-based approach, as the identities of cell-specific PSC molecular markers have remained elusive. This limited approach has precluded our ability to isolate and genetically manipulate PSCs in a cell specific manner. We have identified neuron-glia antigen 2 (NG2) as a unique molecular marker of S100b+ PSCs in skeletal muscle. NG2 is expressed in Schwann cells already associated with the NMJ, indicating that it is a marker of differentiated PSCs. Using a newly generated transgenic mouse in which PSCs are specifically labeled, we show that PSCs have a unique molecular signature that includes genes known to play critical roles in *For correspondence: PSCs and synapses. These findings will serve as a springboard for revealing drivers of PSC [email protected] differentiation and function. -
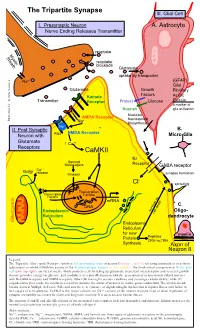
Tripartite Synapse III
The Tripartite Synapse III. Glial Cell I. Presynaptic Neuron A. Astrocyte Nerve Ending Releases Transmitter reuptake Myelin Sheath reuptake blockade Glutamate uptake by transporter Na+ (GFAP) Glial Glutamate Growth Fibrillary Factors Kainate Acidic Transmitter Receptor Protect Glucose Protein a marker of Nourish glia activation Modulate AMPA Receptor Neurosteroid Illustration by: Kendra Scouten Kendra by: Illustration Na+ Biosynthesis Mg++ II. Post Synaptic B. NMDA Receptor Neuron with PSD Micro-Glia Glutamate ↑ Ca++ Receptors CaMKII Bz Second Messengers Receptor GABA receptor ++ Golgi Ca release Kinases synapse formation - Cl sprouting Transcription Transcription Factors Factors mRNA DNA C. Endoplasmic survival vs. Oligo- cell death Reticulum dendrocyte Mitochondria Endoplasmic Reticulum for new myelin Protein Peptides CRH vs.TRH Synthesis Axon of Neuron II Legend: The Tripartite (three-part) Synapse involves: I) a presynaptic axon of neuron I (orange, top left) releasing transmitters to activate (glutamate) or inhibit (GABA-Bz) activity of the II) post-synaptic neuron (yellow, middle). The third critical component is III) the glial cell (pink, top right), an (A) astrocyte which protects cells by taking up glutamate to prevent overexcitation and secretes growth factors; provides energy via glucose; and modulates receptor (R) function with the generation of neurosteroids (which interact with Bz-GABA receptors and NMDA receptors. Other (B) microglia secrete cytokines and scavenge cellular debris; while (C) oligodendrocytes make the myelin necessary to insulate the axons of neurons to insure good conductivity. The myelin sheath breaks down in Multiple Sclerosis (MS) and now there is evidence of oligodendroglia dysfunction in bipolar illness and failure in late stages of schizophrenia. CaMK-II is the major calcium ion (Ca++) sensor of the neuron involved in up or down regulation of synaptic excitability necessary for short and long term memory. -

Tripartite Synapses: Glia, the Unacknowledged Partner
L ETTERS TO THE EDITOR components of the stretch-reflex system that this ‘system’ also includes the major tulated to interconnect and integrate them include: (1) dorsal-root-ganglion cells with sensory and motor systems as well. In a for whatever behavior or function is under their peripheral process that ends in stri- widely used fear-conditioning paradigm, consideration. ated-muscle stretch receptors and their auditory stimuli are used as conditioning Larry Swanson central process that ends on ventral-horn stimuli, foot-shock (somatosensory) stim- Gorica Petrovich motoneurons; and (2) the innervated uli are used as unconditioned stimuli and Neuroscience Program, University of ventral-horn motoneurons themselves. In the behavior of the animal that follows the Southern California, Los Angeles, other words, the stretch-reflex system presentation of such stimuli relies on the CA 90089-2520, USA. itself consists of parts of two classical sys- somatomotor system. In fact, very wide- tems: the somatosensory (proprioceptive) spread parts of the nervous system must References and somatomotor systems. be active during fear conditioning and 1 Lanuza, E., Martínez-Marcos, A. and Martínez-García, F. (1999) Trends Lanuza and colleagues suggest that emotional learning in general. Neurosci. 22, 207 because the basolateral amygdala and cen- Unfortunately, there is no general, 2 Nieuwenhuys, R., ten Donkellar, H.J. tral amygdala are interconnected, and have systematic theory or taxonomy of the and Nicholson, C., eds (1997) The Central Nervous System of Vertebrates, been implicated in fear conditioning and organization of mammalian neural systems. Springer-Verlag emotional learning, these two brain areas The development of one could be a major 3 Swanson, L.W. -

Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis
Review Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis Valentina Cappello 1,* and Maura Francolini 2,* 1 Center for Nanotechnology Innovation@NEST, Istituto Italiano di Tecnologia Piazza San Silvestro 12, 56127 Pisa, Italy 2 Department of Medical Biotechnology and Translational Medicine, Università degli Studi di Milano—Via Vanvitelli 32, 20129 Milano, Italy * Correspondence: [email protected] (V.C.); [email protected] (M.F.); Tel.: +39-050-509123 (V.C.); +39-02-50316977 (M.F.) Received: 28 August 2017; Accepted: 28 September 2017; Published: 3 October 2017 Abstract: Neuromuscular junction assembly and plasticity during embryonic, postnatal, and adult life are tightly regulated by the continuous cross-talk among motor nerve endings, muscle fibers, and glial cells. Altered communications among these components is thought to be responsible for the physiological age-related changes at this synapse and possibly for its destruction in pathological states. Neuromuscular junction dismantling plays a crucial role in the onset of Amyotrophic Lateral Sclerosis (ALS). ALS is characterized by the degeneration and death of motor neurons leading to skeletal muscle denervation, atrophy and, most often, death of the patient within five years from diagnosis. ALS is a non-cell autonomous disease as, besides motor neuron degeneration, glial cells, and possibly muscle fibers, play a role in its onset and progression. Here, we will review the recent literature regarding the mechanisms leading to neuromuscular junction disassembly and muscle denervation focusing on the role of the three players of this peripheral tripartite synapse. Keywords: neuromuscular junction; Amyotrophic Lateral Sclerosis; tripartite synapse 1. The Neuromuscular Junction as a Tripartite Synapse At the end of the 1990, it was proposed that glial cells, besides providing the ideal milieu for neuronal function, might have a role in the modulation of neuronal activity, synaptic neurotransmission, and plasticity. -

Role of Glia in Sculpting Synaptic Connections at the Drosophila Neuromuscular Junction: a Dissertation
University of Massachusetts Medical School eScholarship@UMMS GSBS Dissertations and Theses Graduate School of Biomedical Sciences 2012-01-27 Role of Glia in Sculpting Synaptic Connections at the Drosophila Neuromuscular Junction: A Dissertation Yuly F. Fuentes Medel University of Massachusetts Medical School Let us know how access to this document benefits ou.y Follow this and additional works at: https://escholarship.umassmed.edu/gsbs_diss Part of the Amino Acids, Peptides, and Proteins Commons, Animal Experimentation and Research Commons, Cells Commons, Nervous System Commons, and the Neuroscience and Neurobiology Commons Repository Citation Fuentes Medel YF. (2012). Role of Glia in Sculpting Synaptic Connections at the Drosophila Neuromuscular Junction: A Dissertation. GSBS Dissertations and Theses. https://doi.org/10.13028/ezjw- jx65. Retrieved from https://escholarship.umassmed.edu/gsbs_diss/580 This material is brought to you by eScholarship@UMMS. It has been accepted for inclusion in GSBS Dissertations and Theses by an authorized administrator of eScholarship@UMMS. For more information, please contact [email protected]. ROLE OF GLIA IN SCULPTING SYNAPTIC CONNECTIONS AT THE DROSOPHILA NEUROMUSCULAR JUNCTION A Dissertation Presented By Yuly Fuentes Submitted to the Faculty of the University of Massachusetts Graduate School of Biological Sciences, Worcester in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY January 27th 2012 Program in Neuroscience ii ROLE OF GLIA IN SCULPTING SYNAPTIC CONNECTIONS AT THE DROSOPHILA NEUROMUSCULAR JUNCTION A Dissertation Presented By Yuly Fuentes The signatures of the Dissertation Defense Committee signify completion and approval as to style and content of the Dissertation. _________________________________________ Vivian Budnik, Ph.D. Thesis Advisor _________________________________________ Marc Freeman, Ph.D. -

A Mathematical Model of Tripartite Synapse: Astrocyte Induced Synaptic Plasticity
A Mathematical Model of Tripartite Synapse: Astrocyte Induced Synaptic Plasticity Shivendra Tewari [email protected] Systems Science and Informatics Unit Indian Statistical Institute 8th Mile, Mysore Road Bangalore 560059, India Kaushik Majumdar [email protected] Systems Science and Informatics Unit Indian Statistical Institute 8th Mile, Mysore Road Bangalore 560059, India In this paper we present a mathematical model of tripartite synapses, where astrocytes mediate information flow from the pre-synaptic to the post-synaptic neuron. The model consists of a pre-synaptic bouton, a post-synaptic dendritic spine head, a synaptic cleft and a perisynaptic astrocyte controlling Ca 2+ dynamics inside the synaptic bouton. This in turn controls glutamate release dynamics in the cleft. As a consequence of this, glutamate concentration in the cleft has been modeled, in which glutamate reuptake by astrocytes has also been incorporated. Finally, dendritic spine head dynamics has been modeled. As an application, this model clearly shows synaptic potentiation in the hippocampal region, i.e., astrocyte Ca 2+ mediates synaptic plasticity, which is in conformity with the majority of the recent findings. 1. Introduction One of the most significant challenges in neuroscience is to identify the cellular and molecular processes that underlie learning and memory formation (Lynch, 2004). Cajal originally hypothesized that information storage relies on changes in strength of synaptic connections between neurons that are active (Cajal, 1913). Hebb supported this hypothesis and proposed that if two neurons are active at the same time, the synaptic efficiency of the appropriate synapse will be strengthened (Hebb, 1949). Synaptic transmission is a dynamic process. -

Regulation of Synaptic Development by Astrocyte Signaling Factors and Their Emerging Roles in Substance Abuse
cells Review Regulation of Synaptic Development by Astrocyte Signaling Factors and Their Emerging Roles in Substance Abuse Christopher D. Walker 1, W. Christopher Risher 1 and Mary-Louise Risher 1,2,3,* 1 Department of Biomedical Research, Joan C. Edwards School of Medicine, Huntington, WV 25701, USA; [email protected] (C.D.W.); [email protected] (W.C.R.) 2 Neurobiology Research Laboratory, VA Medical Center, Huntington, WV 25704, USA 3 Department of Psychiatry and Behavioral Sciences, Duke University Medical Center, Durham, NC 27701, USA * Correspondence: [email protected]; Tel.: +1-304-396-3894 Received: 20 December 2019; Accepted: 20 January 2020; Published: 26 January 2020 Abstract: Astrocytes have critical functions throughout the central nervous system (CNS) and have emerged as regulators of synaptic development and function. With their highly complex morphologies, they are able to interact with thousands of synapses via peripheral astrocytic processes (PAPs), ensheathing neuronal axons and dendrites to form the tripartite synapse. In this way, astrocytes engage in crosstalk with neurons to mediate a variety of CNS processes including the regulation of extracellular matrix protein signaling, formation and maintenance of the blood-brain barrier (BBB), axon growth and guidance, homeostasis of the synaptic microenvironment, synaptogenesis, and the promotion of synaptic diversity. In this review, we discuss several key astrocyte signaling factors (thrombospondins, netrins, apolipoproteins, neuregulins, bone morphogenetic proteins, and neuroligins) in the maintenance and regulation of synapse formation. We also explore how these astrocyte signaling factors are impacted by and contribute to substance abuse, particularly alcohol and cocaine use. Keywords: astrocytes; thrombospondin; netrin; apolipoprotein; neuregulins; bone morphogenetic proteins; neuroligin; alcohol use disorder; substance abuse; cocaine 1. -

Program Book
ACNP 53rd Annual Meeting Final Program December 7-11, 2014 JW Marriott Phoenix Desert Ridge Resort and Spa Phoenix, Arizona President: Peter W. Kalivas, Ph.D. Program Committee Chair: Pat R. Levitt, Ph.D. Program Committee Co-Chair: Bita Moghaddam, Ph.D. This meeting is jointly sponsored by the Vanderbilt University School of Medicine Department of Psychiatry and the American College of Neuropsychopharmacology. ACNP Annual Meeting Book Cover 2014.indd 1 10/24/14 8:36 AM Dear Friends and Colleagues Welcome to the 53rd annual meeting of the American College of Neuropsychopharmacology. It has been a great distinction and pleasure for me to work with your colleagues to help develop this year’s program of events and scientific symposia. The JW Marriott is an outstanding venue that promises more than adequate meeting space and areas to gather in discussion offering a great opportunity to have fun, enjoy your colleagues and experience the latest advances in neuroscience discovery related to neuropsychiatric disease. Thanks to the Program Committee and the committee chair, Pat Levitt, and his co-chair, Bita Moghaddam, we have an exciting program for this year’s meeting that contains innovations to promote scientific exchange and provide opportunity to participate across our membership. For example, the evening Workshops that are built around discussion more than presentation are moved into the daytime program. Thanks to the membership’s effort to create a meeting that provides opportunity across our membership, you will experience scientifically excellent symposia that are by far our most demographically diverse. The ACNP is a unique amalgamation of preclinical, clinical, government, academic and industrial researchers. -

THE DANA REVIEW 1 Title: Rethinking the Synapse Author: Brenda Patoine
THE DANA REVIEW http://www.g2conline.info/863 1 Title: Rethinking the Synapse Author: Brenda Patoine In terms of brain function, the synapse is where it all happens. It is at these molecular junctions— 100 trillion or more of them, by current estimates—that neurons talk to one another, exchanging biochemical messages that ultimately orchestrate how we feel, think, remember, and behave. Now, slowly and cautiously, neuroscientists are rewriting the science of the synapse. The classic, textbook view of nerve transmission at the synapse depicts an electrical signal traveling along a nerve fiber and triggering the neuron to release chemical transmitters, which then diffuse across a tiny cleft and lock onto receptors on a neighboring neuron. The nerve signal is thus transmitted from one neuron to the next, like the baton in a relay race. But it is turning out that this tidy, two-party picture may not be telling the whole story. It is increasingly clear that the nonneuronal brain cells called glia are intricately involved in the neuronal crosstalk at synapses. As scientists probe deeper, armed with new experimental techniques, powerful imaging tools, and a better understanding of glia than ever before, a new view of the synapse is emerging. Now Starring: the Astrocyte Central to this new view is the concept of a “tripartite synapse,” in which the astrocyte, a specialized type of glia, is an integral part of the triad. Some scientists are convinced that glia, long shunned as the mere “glue” of the nervous system, as their Greek name implies, in fact play critical, active roles in modulating neuronal transmission. -

Electrical Coupling Between A17 Cells Enhances Reciprocal Inhibitory
www.nature.com/scientificreports Correction: Publisher Correction OPEN Electrical coupling between A17 cells enhances reciprocal inhibitory feedback to rod bipolar cells Received: 9 February 2017 Claudio Elgueta1,2, Felix Leroy 1,3, Alex H. Vielma1,2, Oliver Schmachtenberg1,2 & Accepted: 29 January 2018 Adrian G. Palacios1,2 Published: xx xx xxxx A17 amacrine cells are an important part of the scotopic pathway. Their synaptic varicosities receive glutamatergic inputs from rod bipolar cells (RBC) and release GABA onto the same RBC terminal, forming a reciprocal feedback that shapes RBC depolarization. Here, using patch-clamp recordings, we characterized electrical coupling between A17 cells of the rat retina and report the presence of strongly interconnected and non-coupled A17 cells. In coupled A17 cells, evoked currents preferentially fow out of the cell through GJs and cross-synchronization of presynaptic signals in a pair of A17 cells is correlated to their coupling degree. Moreover, we demonstrate that stimulation of one A17 cell can induce electrical and calcium transients in neighboring A17 cells, thus confrming a functional fow of information through electrical synapses in the A17 coupled network. Finally, blocking GJs caused a strong decrease in the amplitude of the inhibitory feedback onto RBCs. We therefore propose that electrical coupling between A17 cells enhances feedback onto RBCs by synchronizing and facilitating GABA release from inhibitory varicosities surrounding each RBC axon terminal. GJs between A17 cells are therefore critical in shaping the visual fow through the scotopic pathway. Te mammalian retina can elicit behavioral responses afer the detection and processing of just a few photons1. Tis is accomplished by the scotopic pathway, a highly efcient microcircuit formed by distinct sets of dedi- cated excitatory and inhibitory cells2–5.