Astrocytic Regulation of Seizure-Like Behavior
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Abstract Book
Table of Contents Tuesday, May 25, 2021 ............................................................................................................................................................... 5 T1. Astrocyte-Specific Expression of the Extracellular Matrix Gene HtrA1 Regulates Susceptibility to Stress in a Sex- Specific Manner ....................................................................................................................................................................... 5 T2. Plexin-B2 Regulates Migratory Plasticity of Glioblastoma Cells in a 3D-Printed Micropattern Device ............................. 5 T3. Pathoanatomical Mapping of Differential MAPT Expression and Splicing in Progressive Supranuclear Palsy ............... 5 T4. Behavioral Variability in Response to Chronic Stress and Morphine in BXD and Parental Mouse Lines......................... 6 T5. Thyroid-Stimulating Hormone Receptor Regulates Anxiety .............................................................................................. 6 T6. Drugs That Inhibit Microglial Inflammation Also Ameliorate Aβ1-42 Induced Toxicity in C. Elegans ............................... 7 T7. Phosphodiesterase 1b is an Upstream Regulator of a Key Gene Network in the Nucleus Accumbens Driving Addiction- Like Behaviors ......................................................................................................................................................................... 7 T8. Reduced Gap Effect in Children With FOXP1 Syndrome and Autism Spectrum -

Astroglial Networks Scale Synaptic Activity and Plasticity
Astroglial networks scale synaptic activity and plasticity Ulrike Pannascha, Lydia Vargováb,c, Jürgen Reingruberd, Pascal Ezana, David Holcmand, Christian Giaumea, Eva Sykováb,c, and Nathalie Rouacha,1 aCenter for Interdisciplinary Research in Biology, Centre National de la Recherche Scientifique Unité Mixte de Recherche 7241/Institut National de la Santé et de la Recherche Médicale U1050, Collège de France, 75005 Paris, France; bDepartment of Neuroscience and Center for Cell therapy and Tissue Repair, Charles University, Second Faculty of Medicine, 150 06 Prague, Czech Republic; cDepartment of Neuroscience, Institute of Experimental Medicine of the Academy of Sciences of the Czech Republic, 142 20 Prague, Czech Republic; and dInstitut de Biologie de l’Ecole Normale Supérieure, Centre National de la Recherche Scientifique Unité Mixte de Recherche 8197/Institut National de la Santé et de la Recherche Médicale U1024, Department of Computational Biology and Mathematics, Ecole Normale Supérieure, Paris, 75005, France Edited* by Roger A. Nicoll, University of California, San Francisco, CA, and approved April 11, 2011 (received for review November 22, 2010) Astrocytes dynamically interact with neurons to regulate synaptic during synaptic activity. This impairment results in increased transmission. Although the gap junction proteins connexin 30 neuronal excitability, release probability, glutamate spillover, and (Cx30) and connexin 43 (Cx43) mediate the extensive network insertion of postsynaptic AMPARs, unsilencing synapses. Alto- organization of astrocytes, their role in synaptic physiology is un- gether, our findings indicate that gap junction-mediated astroglial known. Here we show, by inactivating Cx30 and Cx43 genes, that networks control synaptic strength by modulating extracellular astroglial networks tone down hippocampal synaptic transmission homeostasis. -

Specific Labeling of Synaptic Schwann Cells Reveals Unique Cellular And
RESEARCH ARTICLE Specific labeling of synaptic schwann cells reveals unique cellular and molecular features Ryan Castro1,2,3, Thomas Taetzsch1,2, Sydney K Vaughan1,2, Kerilyn Godbe4, John Chappell4, Robert E Settlage5, Gregorio Valdez1,2,6* 1Department of Molecular Biology, Cellular Biology, and Biochemistry, Brown University, Providence, United States; 2Center for Translational Neuroscience, Robert J. and Nancy D. Carney Institute for Brain Science and Brown Institute for Translational Science, Brown University, Providence, United States; 3Neuroscience Graduate Program, Brown University, Providence, United States; 4Fralin Biomedical Research Institute at Virginia Tech Carilion, Roanoke, United States; 5Department of Advanced Research Computing, Virginia Tech, Blacksburg, United States; 6Department of Neurology, Warren Alpert Medical School of Brown University, Providence, United States Abstract Perisynaptic Schwann cells (PSCs) are specialized, non-myelinating, synaptic glia of the neuromuscular junction (NMJ), that participate in synapse development, function, maintenance, and repair. The study of PSCs has relied on an anatomy-based approach, as the identities of cell-specific PSC molecular markers have remained elusive. This limited approach has precluded our ability to isolate and genetically manipulate PSCs in a cell specific manner. We have identified neuron-glia antigen 2 (NG2) as a unique molecular marker of S100b+ PSCs in skeletal muscle. NG2 is expressed in Schwann cells already associated with the NMJ, indicating that it is a marker of differentiated PSCs. Using a newly generated transgenic mouse in which PSCs are specifically labeled, we show that PSCs have a unique molecular signature that includes genes known to play critical roles in *For correspondence: PSCs and synapses. These findings will serve as a springboard for revealing drivers of PSC [email protected] differentiation and function. -
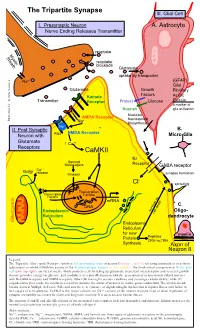
Tripartite Synapse III
The Tripartite Synapse III. Glial Cell I. Presynaptic Neuron A. Astrocyte Nerve Ending Releases Transmitter reuptake Myelin Sheath reuptake blockade Glutamate uptake by transporter Na+ (GFAP) Glial Glutamate Growth Fibrillary Factors Kainate Acidic Transmitter Receptor Protect Glucose Protein a marker of Nourish glia activation Modulate AMPA Receptor Neurosteroid Illustration by: Kendra Scouten Kendra by: Illustration Na+ Biosynthesis Mg++ II. Post Synaptic B. NMDA Receptor Neuron with PSD Micro-Glia Glutamate ↑ Ca++ Receptors CaMKII Bz Second Messengers Receptor GABA receptor ++ Golgi Ca release Kinases synapse formation - Cl sprouting Transcription Transcription Factors Factors mRNA DNA C. Endoplasmic survival vs. Oligo- cell death Reticulum dendrocyte Mitochondria Endoplasmic Reticulum for new myelin Protein Peptides CRH vs.TRH Synthesis Axon of Neuron II Legend: The Tripartite (three-part) Synapse involves: I) a presynaptic axon of neuron I (orange, top left) releasing transmitters to activate (glutamate) or inhibit (GABA-Bz) activity of the II) post-synaptic neuron (yellow, middle). The third critical component is III) the glial cell (pink, top right), an (A) astrocyte which protects cells by taking up glutamate to prevent overexcitation and secretes growth factors; provides energy via glucose; and modulates receptor (R) function with the generation of neurosteroids (which interact with Bz-GABA receptors and NMDA receptors. Other (B) microglia secrete cytokines and scavenge cellular debris; while (C) oligodendrocytes make the myelin necessary to insulate the axons of neurons to insure good conductivity. The myelin sheath breaks down in Multiple Sclerosis (MS) and now there is evidence of oligodendroglia dysfunction in bipolar illness and failure in late stages of schizophrenia. CaMK-II is the major calcium ion (Ca++) sensor of the neuron involved in up or down regulation of synaptic excitability necessary for short and long term memory. -

Tripartite Synapses: Glia, the Unacknowledged Partner
L ETTERS TO THE EDITOR components of the stretch-reflex system that this ‘system’ also includes the major tulated to interconnect and integrate them include: (1) dorsal-root-ganglion cells with sensory and motor systems as well. In a for whatever behavior or function is under their peripheral process that ends in stri- widely used fear-conditioning paradigm, consideration. ated-muscle stretch receptors and their auditory stimuli are used as conditioning Larry Swanson central process that ends on ventral-horn stimuli, foot-shock (somatosensory) stim- Gorica Petrovich motoneurons; and (2) the innervated uli are used as unconditioned stimuli and Neuroscience Program, University of ventral-horn motoneurons themselves. In the behavior of the animal that follows the Southern California, Los Angeles, other words, the stretch-reflex system presentation of such stimuli relies on the CA 90089-2520, USA. itself consists of parts of two classical sys- somatomotor system. In fact, very wide- tems: the somatosensory (proprioceptive) spread parts of the nervous system must References and somatomotor systems. be active during fear conditioning and 1 Lanuza, E., Martínez-Marcos, A. and Martínez-García, F. (1999) Trends Lanuza and colleagues suggest that emotional learning in general. Neurosci. 22, 207 because the basolateral amygdala and cen- Unfortunately, there is no general, 2 Nieuwenhuys, R., ten Donkellar, H.J. tral amygdala are interconnected, and have systematic theory or taxonomy of the and Nicholson, C., eds (1997) The Central Nervous System of Vertebrates, been implicated in fear conditioning and organization of mammalian neural systems. Springer-Verlag emotional learning, these two brain areas The development of one could be a major 3 Swanson, L.W. -

Functional Comparisons of Three Glutamate Transporter Subtypes Cloned from Human Motor Cortex
The Journal of Neuroscience, September 1994, 14(g): 5559-5569 Functional Comparisons of Three Glutamate Transporter Subtypes Cloned from Human Motor Cortex Jeffrey L. Arriza, Wendy A. Fairman, Jacques I. Wadiche, Geoffrey H. Murdoch, Michael P. Kavanaugh, and Susan G. Amara The Vellum Institute for Advanced Biomedical Research, Oregon Health Sciences University, Portland, Oregon 97201 Reuptake plays an important role in regulating synaptic and peripheral tissues(Christensen, 1990) and in the nervous system extracellular concentrations of glutamate. Three glutamate (Kanner and Schuldiner, 1987; Nicholls and Attwell, 1990). transporters expressed in human motor cortex, termed Transport serves a special function in the brain by mediating EAATl , EAATP, and EAAT3 (for excitatory amino acid trans- the reuptake of glutamate releasedat excitatory synapses.Glu- porter), have been characterized by their molecular cloning tamate can be reaccumulated from the synaptic cleft by the and functional expression. Each EAAT subtype mRNA was presynaptic nerve terminal (Eliasof and Werblin, 1993) or by found in all human brain regions analyzed. The most prom- glial uptake of transmitter diffusing from the cleft (Nicholls and inent regional variation in message content was in cerebel- Attwell, 1990; Schwartz and Tachibana, 1990; Barbour et al., lum where EAATl expression predominated. EAATl and 199 1). The activities of neuronal and glial transporters influence EAAT3 mRNAs were also expressed in various non-nervous the temporal and spatial dynamics of the chemical signal in tissues, whereas expression of EAATS was largely restricted other neurotransmitter systems(Hille, 1992; Bruns et al., 1993; to brain. The kinetic parameters and pharmacological char- Isaacsonet al., 1993), but such effects have not yet been dem- acteristics of transport mediated by each EAAT subtype onstrated at glutamatergic synapses(Hestrin et al., 1990; Sar- were determined in transfected mammalian cells by radio- antis et al., 1993). -

OCD Candidate Gene SLC1A1/EAAT3 Impacts Basal Ganglia-Mediated Activity and Stereotypic Behavior
OCD candidate gene SLC1A1/EAAT3 impacts basal ganglia-mediated activity and stereotypic behavior Isaac D. Zikea, Muhammad O. Chohanb, Jared M. Kopelmanc,d,e, Emily N. Krasnowb, Daniel Flickerf,g,h,i, Katherine M. Nautiyalj,k, Michael Bubserl, Christoph Kellendonkm,n,o,p, Carrie K. Jonesa,l, Gregg Stanwoodq, Kenji Fransis Tanakar, Holly Mooreb, Susanne E. Ahmaric,d,e,1, and Jeremy Veenstra-VanderWeeleb,m,n,1 aDepartment of Pharmacology, Vanderbilt University Medical Center, Nashville, TN 37232; bNew York State Psychiatric Institute, New York, NY 10032; cDepartment of Psychiatry, University of Pittsburgh, Pittsburgh, PA 15260; dCenter for Neuroscience Program, University of Pittsburgh, Pittsburgh, PA 15260; eCenter for the Neural Basis of Cognition, University of Pittsburgh, Pittsburgh, PA 15260; fHoward Hughes Medical Institute, Department of Molecular Biology, Massachusetts General Hospital, Boston, MA 02114; gCenter for Human Genetic Research, Massachusetts General Hospital, Boston, MA 02114; hDepartment of Systems Biology, Harvard Medical School, Boston, MA 02115; iBroad Institute, Cambridge, MA 02142; jDepartment of Psychiatry, Columbia University Medical Center, New York, NY 10032; kDivision of Integrative Neuroscience, New York State Psychiatric Institute, New York, NY 10032; lVanderbilt Center for Neuroscience Drug Discovery, Vanderbilt University Medical Center, Nashville, TN 37232; mDepartment of Psychiatry, Columbia University Medical Center, New York, NY 10032; nSackler Institute for Developmental Psychobiology, Columbia University Medical Center, New York, NY 10032; oDepartment of Anesthesiology, Columbia University Medical Center, New York, NY 10032; pDivision of Molecular Therapeutics, New York State Psychiatric Institute, New York, NY 10032; qDepartment of Biomedical Sciences, Florida State University College of Medicine, Tallahassee, FL 32304; and rDepartment of Neuropsychiatry, School of Medicine, Keio University, Tokyo 108-8345, Japan Edited by Susan G. -

Neuronal and Glial Mechanisms Underlying BBB Dysfunction-Induced Epileptogenesis
Neuronal and Glial Mechanisms Underlying BBB Dysfunction-Induced Epileptogenesis Thesis submitted in partial fulfillment of the requirements for the degree of “DOCTOR OF PHILOSOPHY” by Yaron David Submitted to the Senate of Ben-Gurion University of the Negev November 2011 Beer-Sheva Neuronal and Glial Mechanisms Underlying BBB Dysfunction-Induced Epileptogenesis Thesis submitted in partial fulfillment of the requirements for the degree of “DOCTOR OF PHILOSOPHY” by Yaron David Submitted to the Senate of Ben-Gurion University of the Negev Approved by the advisor_________ Approved by the Dean of the Kreitman School of Advanced Graduate Studies_______ November 2011 Beer-Sheva This work was carried out under the supervision of Professor Alon Friedman The department of Physiology, Faculty of Health Sciences Ben-Gurion University of the Negev. First and foremost, I would like to thank my mentor, Prof. Alon Friedman, who offered an opportunity to a young student who knew absolutely nothing in the field of neuroscience. Your enthusiasm for the pursuit of knowledge is contagious. I also thank you for being a friend. I would like to thanks all those people I encountered throughout the years: Oren Tomkins who brought me to the lab. Uwe Heinemann who kindly opened the gates of the Institute fur Physiologie, in Berlin for me. Sebastian Ivens, for teaching me electrophysiology and being a good friend. Daniela Kaufer and Luisa P. Flores from UC Berkley, without whom I am sure I wouldn‘t have any molecular studies to present. Ilya Fleidervish, with whom every hour is like a semester of teaching. Maya Ketzef, a good friend which never ceases to help me and others. -

Myo-Inositol Supplementation Augments Visual System Maturation in Mice
Myo-Inositol Supplementation Augments Visual System Maturation in Mice A thesis submitted by Seth William Vogel in partial fulfillment of the requirements for the degree of PhD in Neuroscience Tufts University Graduate School of Biomedical Sciences February 2020 Advisor: Thomas Biederer, PhD Abstract Aside from macronutrients, breast milk contains hundreds of micronutrients including vitamins, minerals, and bioactive proteins. Though it is almost universally acknowledged that breast milk is the best source of infant nutrition, the precise contributions of various milk nutrients towards human development is incompletely understood. Myo-inositol is a six-carbon polyol conserved in breast milk across mammals that is thought to play a role in brain development. Using the mouse visual system as a model, I found that postnatal dietary supplementation of myo-inositol increases segregation of eye inputs in the lateral geniculate nucleus of the thalamus. In addition, the number of presynaptic inputs engulfed by thalamic microglia was significantly increased. Microglia morphology was unchanged, suggesting no change in activation state. Instead my data are consistent with inositol supporting the selective elimination of retinal inputs that underlies LGN retinotopic refinement and input segregation. In the primary visual cortex of adult animals I further found a specific increase in the size of Homer+ excitatory post-synaptic puncta. The size of other synaptic markers was not affected. These changes were accompanied by a significant enhancement of visual acuity in awake and behaving adult animals that had been treated with inositol. To the best of my knowledge no other dietary factors are known to increase microglial engulfment of retinogeniculate terminals, enhance segregation in the LGN, or enlarge post-synaptic specializations. -

Glutamate Transport - Relative Rates of Net Uptake and Heteroexchange
Glutamate transport - relative rates of net uptake and heteroexchange by Zhou Yun 周 云 Master thesis Programme for Cell Biology Department of Molecular Biosciences University of Oslo 1 Table of contents TABLE OF CONTENTS ABSTRACT ACKNOWLEDGEMENTS ABBREVIATIONS INTRODUCTION METABOLISM OF GLUTAMATE GLUTAMATE TRANSPORTERS GLUTAMATE TRANSPORTER STRUCTURE MECHANISM OF GLUTAMATE UPTAKE ANION CONDUCTANCE IN GLUTAMATE TRANSPORTERS TWO DIFFERENT MODES OF SUBSTRATE TRANSLOCATION (EXCHANGE AND NET UPTAKE) WHICH EAAT-SUBTYPE AND WHICH CELLULAR COMPONENT IS RESPONSIBLE FOR MOST BRAIN GLUTAMATE UPTAKE MATERIALS AND METHODS MATERIALS ANIMALS GEL FILTRATION PREPARATION OF RECONSITUTION MIXTURE RECONSTITUTION OF GLUTAMATE TRANSPORTERS INTO LIPOSOMES UPTAKE REACTION FOR RADIOACTIVE AMINO ACID FLUORESCENCE MEASUREMENT RESULTS TEST OF RECONSITUTED TRANSPORTERS UNDER CONDITIONS FAVOURING NET UPTAKE OR HETEROCHANGE LEAKAGE OF GLUTAMATE FROM THE LIPOSOMES UNDERESTIMATION OF THE RELATIVE RATE OF NET UPTAKE IMPORTANCE OF ANIONS D-ASPARTATE VERSUS L-GLUTAMATE RATES OF EXCHANGE AND NET UPTAKE AT SHORTER INCUBATION TIMES AND HIGHER EXTERNAL SUBSTRATE CONCENTRATIONS EFFECTS OF PCBS AND ARACHIDONIC ACID ON TRANSPORTER FUNCTION 2 DISCUSSION THE ADVANTAGE OF THE LIPOSOME ASSAY THE INTEGRITY OF THE LIPOSOME WITH RESPECT TO GLUTAMATE TRANSPORT-ASSOCIATED CHARGE TRANSFER AFFECTS NET UPTAKE AND EXCHANGE DIFFERENTLY THE IMPORTANCE OF THE COMPOSITION OF THE LIPID MEMBRANE FOR TRANSFORTER FUNCTION WHY TERMINALS IN HIPPOCAMPAL SLICES TAKE UP AS MUCH EXTERNAL SUBSTRATE AS GLIA DURING IN VITRO INCUBATION WITH SUBSTRATE IN SPITE OF FEWER TRANSPORTERS CONCLUSION REFERENCES 3 Abstract Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system (CNS), and is inactivated by cellular uptake, mostly catalyzed by glutamate (excitatory amino acid) transporter subtype number 2 (EAAT2). -

Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis
Review Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis Valentina Cappello 1,* and Maura Francolini 2,* 1 Center for Nanotechnology Innovation@NEST, Istituto Italiano di Tecnologia Piazza San Silvestro 12, 56127 Pisa, Italy 2 Department of Medical Biotechnology and Translational Medicine, Università degli Studi di Milano—Via Vanvitelli 32, 20129 Milano, Italy * Correspondence: [email protected] (V.C.); [email protected] (M.F.); Tel.: +39-050-509123 (V.C.); +39-02-50316977 (M.F.) Received: 28 August 2017; Accepted: 28 September 2017; Published: 3 October 2017 Abstract: Neuromuscular junction assembly and plasticity during embryonic, postnatal, and adult life are tightly regulated by the continuous cross-talk among motor nerve endings, muscle fibers, and glial cells. Altered communications among these components is thought to be responsible for the physiological age-related changes at this synapse and possibly for its destruction in pathological states. Neuromuscular junction dismantling plays a crucial role in the onset of Amyotrophic Lateral Sclerosis (ALS). ALS is characterized by the degeneration and death of motor neurons leading to skeletal muscle denervation, atrophy and, most often, death of the patient within five years from diagnosis. ALS is a non-cell autonomous disease as, besides motor neuron degeneration, glial cells, and possibly muscle fibers, play a role in its onset and progression. Here, we will review the recent literature regarding the mechanisms leading to neuromuscular junction disassembly and muscle denervation focusing on the role of the three players of this peripheral tripartite synapse. Keywords: neuromuscular junction; Amyotrophic Lateral Sclerosis; tripartite synapse 1. The Neuromuscular Junction as a Tripartite Synapse At the end of the 1990, it was proposed that glial cells, besides providing the ideal milieu for neuronal function, might have a role in the modulation of neuronal activity, synaptic neurotransmission, and plasticity. -

Role of Glia in Sculpting Synaptic Connections at the Drosophila Neuromuscular Junction: a Dissertation
University of Massachusetts Medical School eScholarship@UMMS GSBS Dissertations and Theses Graduate School of Biomedical Sciences 2012-01-27 Role of Glia in Sculpting Synaptic Connections at the Drosophila Neuromuscular Junction: A Dissertation Yuly F. Fuentes Medel University of Massachusetts Medical School Let us know how access to this document benefits ou.y Follow this and additional works at: https://escholarship.umassmed.edu/gsbs_diss Part of the Amino Acids, Peptides, and Proteins Commons, Animal Experimentation and Research Commons, Cells Commons, Nervous System Commons, and the Neuroscience and Neurobiology Commons Repository Citation Fuentes Medel YF. (2012). Role of Glia in Sculpting Synaptic Connections at the Drosophila Neuromuscular Junction: A Dissertation. GSBS Dissertations and Theses. https://doi.org/10.13028/ezjw- jx65. Retrieved from https://escholarship.umassmed.edu/gsbs_diss/580 This material is brought to you by eScholarship@UMMS. It has been accepted for inclusion in GSBS Dissertations and Theses by an authorized administrator of eScholarship@UMMS. For more information, please contact [email protected]. ROLE OF GLIA IN SCULPTING SYNAPTIC CONNECTIONS AT THE DROSOPHILA NEUROMUSCULAR JUNCTION A Dissertation Presented By Yuly Fuentes Submitted to the Faculty of the University of Massachusetts Graduate School of Biological Sciences, Worcester in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY January 27th 2012 Program in Neuroscience ii ROLE OF GLIA IN SCULPTING SYNAPTIC CONNECTIONS AT THE DROSOPHILA NEUROMUSCULAR JUNCTION A Dissertation Presented By Yuly Fuentes The signatures of the Dissertation Defense Committee signify completion and approval as to style and content of the Dissertation. _________________________________________ Vivian Budnik, Ph.D. Thesis Advisor _________________________________________ Marc Freeman, Ph.D.